

Characterization of functional methylomes by next-generation capture sequencing identifies novel disease-associated variants
- PMID: 26021296
- PMCID: PMC4544751
- DOI: 10.1038/ncomms8211
Characterization of functional methylomes by next-generation capture sequencing identifies novel disease-associated variants
Erratum in
-
Erratum: Characterization of functional methylomes by next-generation capture sequencing identifies novel disease-associated variants.Nat Commun. 2015 Jul 29;6:8016. doi: 10.1038/ncomms9016. Nat Commun. 2015. PMID: 26219997 Free PMC article. No abstract available.
Abstract
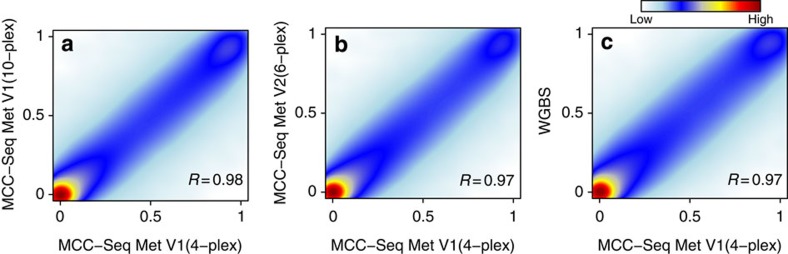
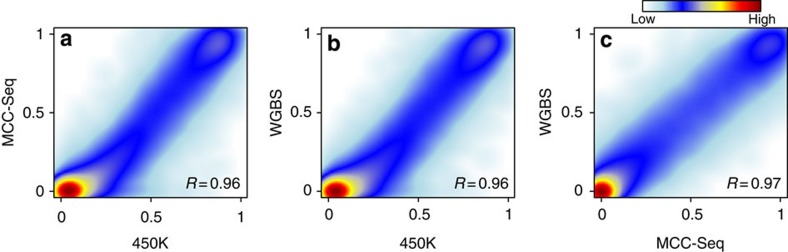
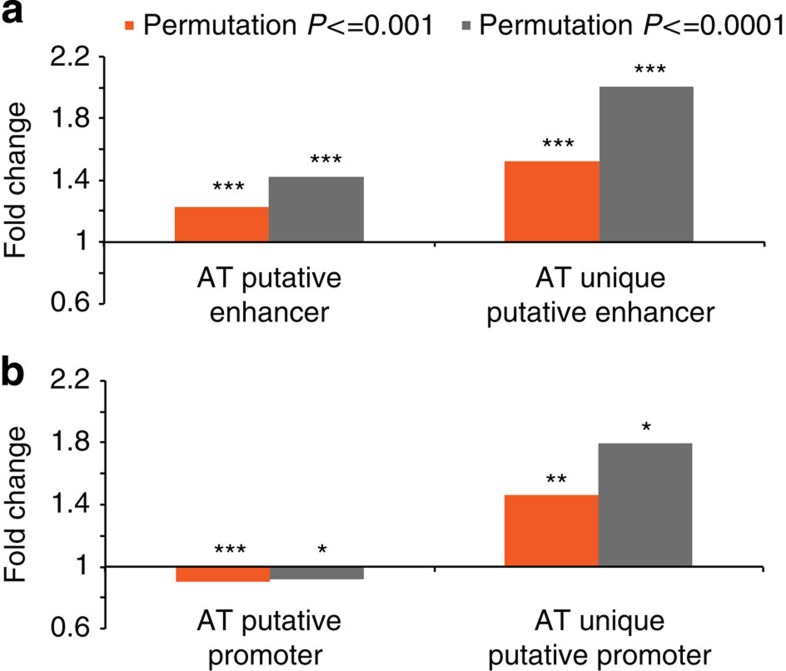
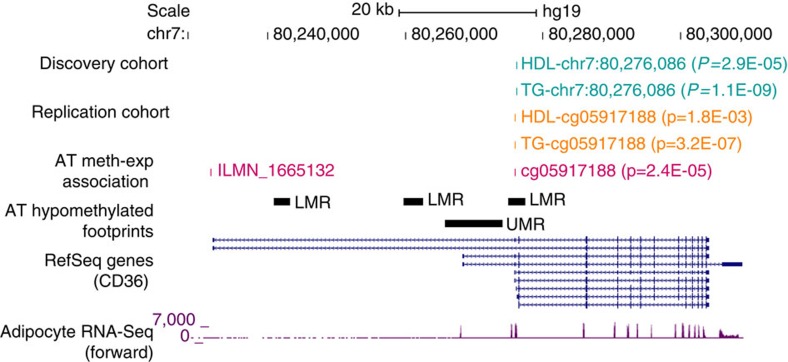
Most genome-wide methylation studies (EWAS) of multifactorial disease traits use targeted arrays or enrichment methodologies preferentially covering CpG-dense regions, to characterize sufficiently large samples. To overcome this limitation, we present here a new customizable, cost-effective approach, methylC-capture sequencing (MCC-Seq), for sequencing functional methylomes, while simultaneously providing genetic variation information. To illustrate MCC-Seq, we use whole-genome bisulfite sequencing on adipose tissue (AT) samples and public databases to design AT-specific panels. We establish its efficiency for high-density interrogation of methylome variability by systematic comparisons with other approaches and demonstrate its applicability by identifying novel methylation variation within enhancers strongly correlated to plasma triglyceride and HDL-cholesterol, including at CD36. Our more comprehensive AT panel assesses tissue methylation and genotypes in parallel at ∼4 and ∼3 M sites, respectively. Our study demonstrates that MCC-Seq provides comparable accuracy to alternative approaches but enables more efficient cataloguing of functional and disease-relevant epigenetic and genetic variants for large-scale EWAS.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Jones P. A. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 13, 484–492 (2012). - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
