

The syndrome of hemophagocytic lymphohistiocytosis in primary immunodeficiencies: implications for differential diagnosis and pathogenesis
- PMID: 26022711
- PMCID: PMC4486233
- DOI: 10.3324/haematol.2014.121608
The syndrome of hemophagocytic lymphohistiocytosis in primary immunodeficiencies: implications for differential diagnosis and pathogenesis
Abstract
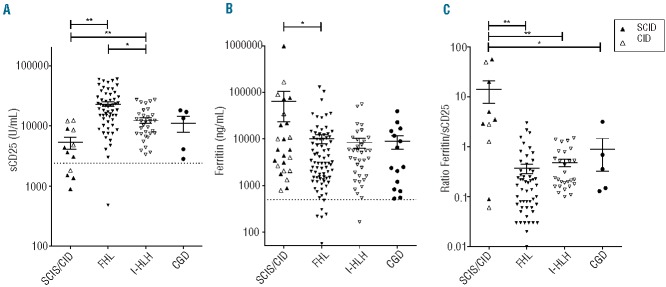
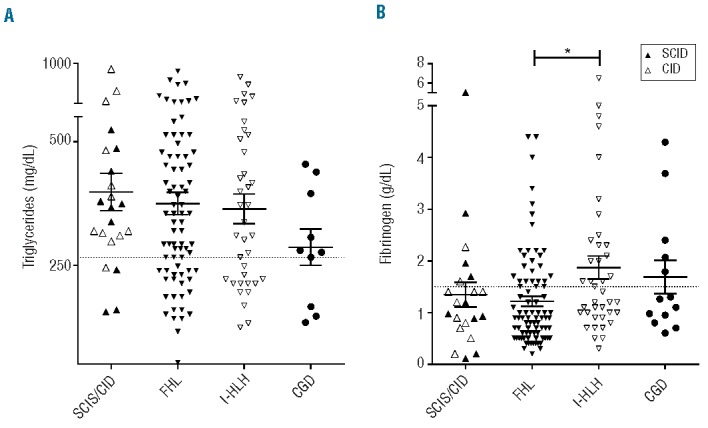
Hemophagocytic lymphohistiocytosis is a hyperinflammatory syndrome defined by clinical and laboratory criteria. Current criteria were created to identify patients with familial hemophagocytic lmyphohistiocytosis in immediate need of immunosuppressive therapy. However, these criteria also identify patients with infection-associated hemophagocytic inflammatory states lacking genetic defects typically predisposing to hemophagocytic lymphohistiocytosis. These patients include those with primary immunodeficiencies, in whom the pathogenesis of the inflammatory syndrome may be distinctive and aggressive immunosuppression is contraindicated. To better characterize hemophagocytic inflammation associated with immunodeficiencies, we combined an international survey with a literature search and identified 63 patients with primary immunodeficiencies other than cytotoxicity defects or X-linked lymphoproliferative disorders, presenting with conditions fulfilling current criteria for hemophagocytic lymphohistiocytosis. Twelve patients had severe combined immunodeficiency with <100/μL T cells, 18 had partial T-cell deficiencies; episodes of hemophagocytic lymphohistiocytosis were mostly associated with viral infections. Twenty-two patients had chronic granulomatous disease with hemophagocytic episodes mainly associated with bacterial infections. Compared to patients with cytotoxicity defects, patients with T-cell deficiencies had lower levels of soluble CD25 and higher ferritin concentrations. Other criteria for hemophagocytoc lymphohistiocytosis were not discriminative. Thus: (i) a hemophagocytic inflammatory syndrome fulfilling criteria for hemophagocytic lymphohistiocytosis can be the initial manifestation of primary immunodeficiencies; (ii) this syndrome can develop despite severe deficiency of T and NK cells, implying that the pathophysiology is distinct and not appropriately described as "lympho"-histiocytosis in these patients; and (iii) current criteria for hemophagocytoc lymphohistiocytosis are insufficient to differentiate hemophagocytic inflammatory syndromes with different pathogeneses. This is important because of implications for therapy, in particular for protocols targeting T cells.
Copyright© Ferrata Storti Foundation.
Figures
References
-
- Janka GE, Lehmberg K. Hemophagocytic syndromes - an update. Blood Rev. 2014;28(4):135–142. - PubMed
-
- Pachlopnik Schmid J, Cote M, Menager MM, et al. Inherited defects in lymphocyte cytotoxic activity. Immunol Rev. 2010;235(1):10–23. - PubMed
-
- Jordan MB, Hildeman D, Kappler J, Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood. 2004;104(3):735–743. - PubMed
-
- Risma K, Jordan MB. Hemophagocytic lymphohistiocytosis: updates and evolving concepts. Curr Opin Pediatr. 2012;24(1):9–15. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
