

Airway epithelial cell PPARγ modulates cigarette smoke-induced chemokine expression and emphysema susceptibility in mice
- PMID: 26024894
- PMCID: PMC4525123
- DOI: 10.1152/ajplung.00287.2014
Airway epithelial cell PPARγ modulates cigarette smoke-induced chemokine expression and emphysema susceptibility in mice
Abstract
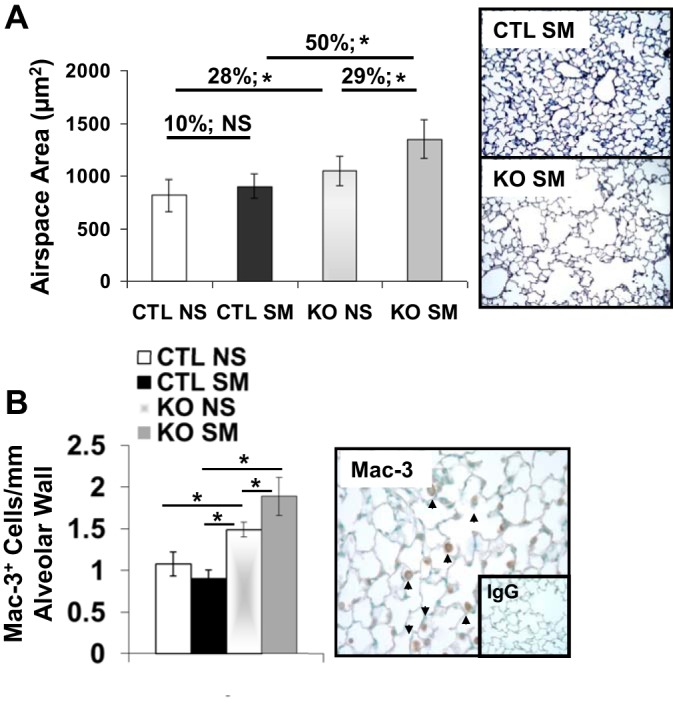
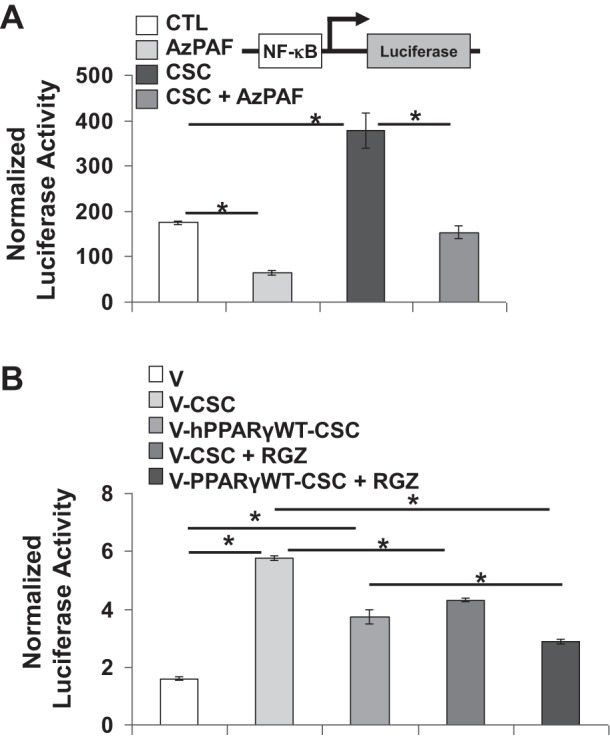
Chronic obstructive pulmonary disease (COPD) is a highly prevalent, chronic inflammatory lung disease with limited existing therapeutic options. While modulation of peroxisome proliferator-activating receptor (PPAR)-γ activity can modify inflammatory responses in several models of lung injury, the relevance of the PPARG pathway in COPD pathogenesis has not been previously explored. Mice lacking Pparg specifically in airway epithelial cells displayed increased susceptibility to chronic cigarette smoke (CS)-induced emphysema, with excessive macrophage accumulation associated with increased expression of chemokines, Ccl5, Cxcl10, and Cxcl15. Conversely, treatment of mice with a pharmacological PPARγ activator attenuated Cxcl10 and Cxcl15 expression and macrophage accumulation in response to CS. In vitro, CS increased lung epithelial cell chemokine expression in a PPARγ activation-dependent fashion. The ability of PPARγ to regulate CS-induced chemokine expression in vitro was not specifically associated with peroxisome proliferator response element (PPRE)-mediated transactivation activity but was correlated with PPARγ-mediated transrepression of NF-κB activity. Pharmacological or genetic activation of PPARγ activity abrogated CS-dependent induction of NF-κB activity. Regulation of NF-κB activity involved direct PPARγ-NF-κB interaction and PPARγ-mediated effects on IKK activation, IκBα degradation, and nuclear translocation of p65. Our data indicate that PPARG represents a disease-relevant pathophysiological and pharmacological target in COPD. Its activation state likely contributes to NF-κB-dependent, CS-induced chemokine-mediated regulation of inflammatory cell accumulation.
Keywords: chronic obstructive pulmonary disease; lung inflammation; nuclear factor-kB; peroxisome proliferator-activating receptor-γ; rosiglitazone.
Figures





Similar articles
-
Down-regulated peroxisome proliferator-activated receptor γ (PPARγ) in lung epithelial cells promotes a PPARγ agonist-reversible proinflammatory phenotype in chronic obstructive pulmonary disease (COPD).J Biol Chem. 2014 Mar 7;289(10):6383-6393. doi: 10.1074/jbc.M113.536805. Epub 2013 Dec 24. J Biol Chem. 2014. Retraction in: J Biol Chem. 2019 Jan 4;294(1):69. doi: 10.1074/jbc.RX118.007042. PMID: 24368768 Free PMC article. Retracted.
-
Rosiglitazone attenuates the metalloprotease/anti-metalloprotease imbalance in emphysema induced by cigarette smoke: involvement of extracellular signal-regulated kinase and NFκB signaling.Int J Chron Obstruct Pulmon Dis. 2015 Apr 7;10:715-24. doi: 10.2147/COPD.S77514. eCollection 2015. Int J Chron Obstruct Pulmon Dis. 2015. PMID: 25897215 Free PMC article.
-
Up-regulation of PPAR-γ involved in the therapeutic effect of icariin on cigarette smoke-induced inflammation.Pulm Pharmacol Ther. 2023 Apr;79:102197. doi: 10.1016/j.pupt.2023.102197. Epub 2023 Jan 20. Pulm Pharmacol Ther. 2023. PMID: 36690317
-
Curcumin inhibits cigarette smoke-induced inflammation via modulating the PPARγ-NF-κB signaling pathway.Food Funct. 2019 Dec 11;10(12):7983-7994. doi: 10.1039/c9fo02159k. Food Funct. 2019. PMID: 31773117
-
Deacetylases and NF-kappaB in redox regulation of cigarette smoke-induced lung inflammation: epigenetics in pathogenesis of COPD.Antioxid Redox Signal. 2008 Apr;10(4):799-811. doi: 10.1089/ars.2007.1938. Antioxid Redox Signal. 2008. PMID: 18220485 Free PMC article. Review.
Cited by
-
Inhibition of NADPH Oxidase-Derived Reactive Oxygen Species Decreases Expression of Inflammatory Cytokines in A549 Cells.Inflammation. 2019 Dec;42(6):2205-2214. doi: 10.1007/s10753-019-01084-0. Inflammation. 2019. PMID: 31612365 Free PMC article.
-
Scleroderma keratinocytes promote fibroblast activation independent of transforming growth factor beta.Rheumatology (Oxford). 2017 Nov 1;56(11):1970-1981. doi: 10.1093/rheumatology/kex280. Rheumatology (Oxford). 2017. PMID: 28968684 Free PMC article.
-
Nicotine Modulates Growth Factors and MicroRNA to Promote Inflammatory and Fibrotic Processes.J Pharmacol Exp Ther. 2019 Feb;368(2):169-178. doi: 10.1124/jpet.118.252650. Epub 2018 Nov 16. J Pharmacol Exp Ther. 2019. PMID: 30446578 Free PMC article.
-
ATF1 and miR-27b-3p drive intervertebral disc degeneration through the PPARG/NF-κB signaling axis.Commun Biol. 2025 May 14;8(1):751. doi: 10.1038/s42003-025-08186-6. Commun Biol. 2025. PMID: 40369110 Free PMC article.
-
Ribosomal Protein S3 Gene Silencing Protects Against Cigarette Smoke-Induced Acute Lung Injury.Mol Ther Nucleic Acids. 2018 Sep 7;12:370-380. doi: 10.1016/j.omtn.2018.05.027. Epub 2018 Jul 3. Mol Ther Nucleic Acids. 2018. PMID: 30195775 Free PMC article.
References
-
- Akiyama TE, Sakai S, Lambert G, Nicol CJ, Matsusue K, Pimprale S, Lee YH, Ricote M, Glass CK, Brewer HB Jr, Gonzalez FJ. Conditional disruption of the peroxisome proliferator-activated receptor gamma gene in mice results in lowered expression of ABCA1, ABCG1, and apoE in macrophages and reduced cholesterol efflux. Mol Cell Biol 22: 2607–2619, 2002. - PMC - PubMed
-
- Amoruso A, Bardelli C, Gunella G, Fresu LG, Ferrero V, Brunelleschi S. Quantification of PPAR-gamma protein in monocyte/macrophages from healthy smokers and non-smokers: a possible direct effect of nicotine. Life Sci 81: 906–915, 2007. - PubMed
-
- Anto RJ, Mukhopadhyay A, Shishodia S, Gairola CG, Aggarwal BB. Cigarette smoke condensate activates nuclear transcription factor-kappaB through phosphorylation and degradation of IkappaB(alpha): correlation with induction of cyclooxygenase-2. Carcinogenesis 23: 1511–1518, 2002. - PubMed
-
- Aoshiba K, Yokohori N, Nagai A. Alveolar wall apoptosis causes lung destruction and emphysematous changes. Am J Respir Cell Mol Biol 28: 555–562, 2003. - PubMed
-
- Bailey ST, Ghosh S. ‘PPAR’ting ways with inflammation. Nat Immunol 6: 966–967, 2005. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases