

Alteration of ornithine metabolism leads to dominant and recessive hereditary spastic paraplegia
- PMID: 26026163
- PMCID: PMC4553756
- DOI: 10.1093/brain/awv143
Alteration of ornithine metabolism leads to dominant and recessive hereditary spastic paraplegia
Abstract
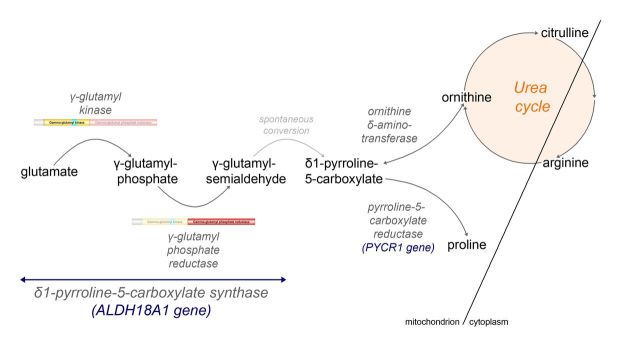
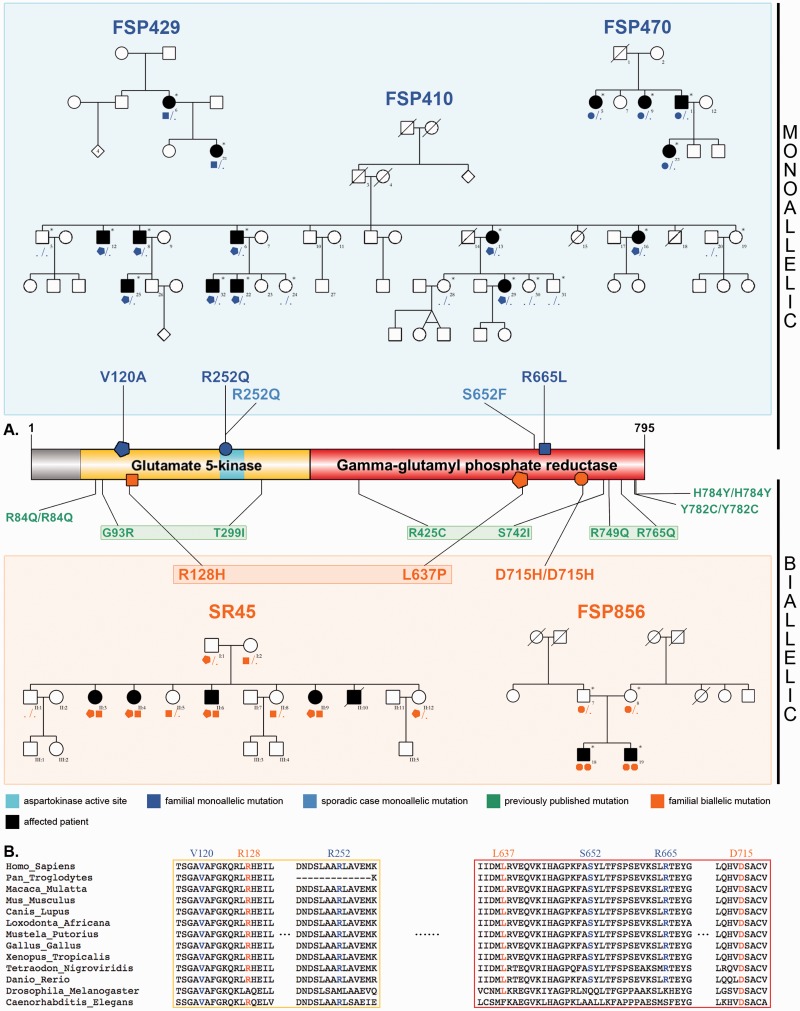
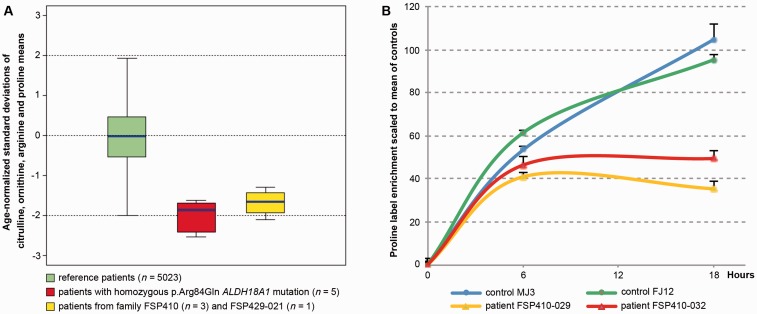
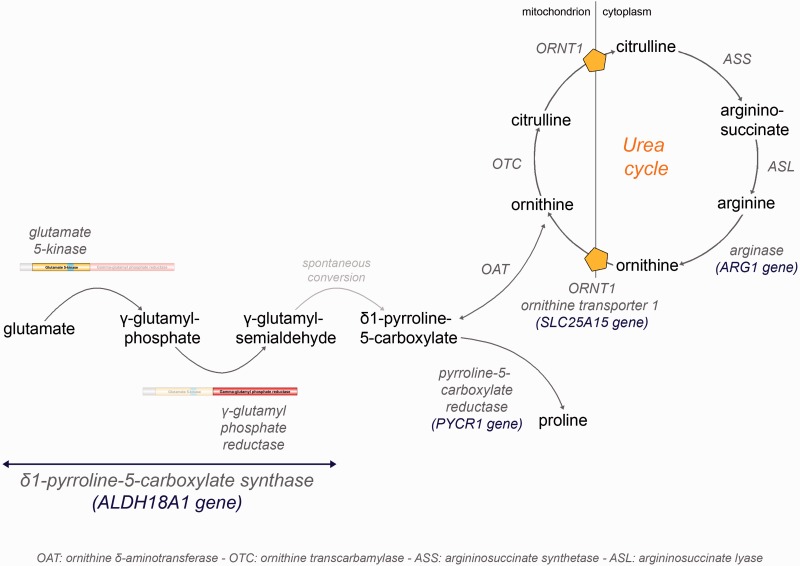
Hereditary spastic paraplegias are heterogeneous neurological disorders characterized by a pyramidal syndrome with symptoms predominantly affecting the lower limbs. Some limited pyramidal involvement also occurs in patients with an autosomal recessive neurocutaneous syndrome due to ALDH18A1 mutations. ALDH18A1 encodes delta-1-pyrroline-5-carboxylate synthase (P5CS), an enzyme that catalyses the first and common step of proline and ornithine biosynthesis from glutamate. Through exome sequencing and candidate gene screening, we report two families with autosomal recessive transmission of ALDH18A1 mutations, and predominant complex hereditary spastic paraplegia with marked cognitive impairment, without any cutaneous abnormality. More interestingly, we also identified monoallelic ALDH18A1 mutations segregating in three independent families with autosomal dominant pure or complex hereditary spastic paraplegia, as well as in two sporadic patients. Low levels of plasma ornithine, citrulline, arginine and proline in four individuals from two families suggested P5CS deficiency. Glutamine loading tests in two fibroblast cultures from two related affected subjects confirmed a metabolic block at the level of P5CS in vivo. Besides expanding the clinical spectrum of ALDH18A1-related pathology, we describe mutations segregating in an autosomal dominant pattern. The latter are associated with a potential trait biomarker; we therefore suggest including amino acid chromatography in the clinico-genetic work-up of hereditary spastic paraplegia, particularly in dominant cases, as the associated phenotype is not distinct from other causative genes.
Keywords: ALDH18A1; citrulline; delta-1-pyrroline-5-carboxylate synthase; hereditary spastic paraplegia; ornithine.
© The Author (2015). Published by Oxford University Press on behalf of the Guarantors of Brain. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
Comment in
-
Reply: ALDH18A1 gene mutations cause dominant spastic paraplegia SPG9: loss of function effect and plausibility of a dominant negative mechanism.Brain. 2016 Jan;139(Pt 1):e4. doi: 10.1093/brain/awv248. Epub 2015 Aug 21. Brain. 2016. PMID: 26297557 No abstract available.
-
ALDH18A1 gene mutations cause dominant spastic paraplegia SPG9: loss of function effect and plausibility of a dominant negative mechanism.Brain. 2016 Jan;139(Pt 1):e3. doi: 10.1093/brain/awv247. Epub 2015 Aug 21. Brain. 2016. PMID: 26297558 No abstract available.
Similar articles
-
Compound heterozygous mutations in two different domains of ALDH18A1 do not affect the amino acid levels in a patient with hereditary spastic paraplegia.Neurogenetics. 2018 Aug;19(3):145-149. doi: 10.1007/s10048-018-0547-7. Epub 2018 May 12. Neurogenetics. 2018. PMID: 29754261
-
Tremor as an early sign of hereditary spastic paraplegia due to mutations in ALDH18A1.Brain Dev. 2021 Jan;43(1):144-151. doi: 10.1016/j.braindev.2020.07.015. Epub 2020 Aug 11. Brain Dev. 2021. PMID: 32798076
-
Δ1 -Pyrroline-5-carboxylate synthetase deficiency: An emergent multifaceted urea cycle-related disorder.J Inherit Metab Dis. 2020 Jul;43(4):657-670. doi: 10.1002/jimd.12220. Epub 2020 Feb 9. J Inherit Metab Dis. 2020. PMID: 32017139 Review.
-
SPG9A with the new occurrence of an ALDH18A1 mutation in a CMT1A family with PMP22 duplication: case report.BMC Neurol. 2021 Feb 11;21(1):64. doi: 10.1186/s12883-021-02087-x. BMC Neurol. 2021. PMID: 33573605 Free PMC article.
-
Hereditary Spastic Paraplegia Is a Common Phenotypic Finding in ARG1 Deficiency, P5CS Deficiency and HHH Syndrome: Three Inborn Errors of Metabolism Caused by Alteration of an Interconnected Pathway of Glutamate and Urea Cycle Metabolism.Front Neurol. 2019 Feb 22;10:131. doi: 10.3389/fneur.2019.00131. eCollection 2019. Front Neurol. 2019. PMID: 30853934 Free PMC article. Review.
Cited by
-
Hereditary Spastic Paraplegia: Clinical and Genetic Hallmarks.Cerebellum. 2017 Apr;16(2):525-551. doi: 10.1007/s12311-016-0803-z. Cerebellum. 2017. PMID: 27271711 Review.
-
Lipodystrophy-associated progeroid syndromes.Hormones (Athens). 2022 Dec;21(4):555-571. doi: 10.1007/s42000-022-00386-7. Epub 2022 Jul 15. Hormones (Athens). 2022. PMID: 35835948 Review.
-
Recurrent De Novo Mutations Affecting Residue Arg138 of Pyrroline-5-Carboxylate Synthase Cause a Progeroid Form of Autosomal-Dominant Cutis Laxa.Am J Hum Genet. 2015 Sep 3;97(3):483-92. doi: 10.1016/j.ajhg.2015.08.001. Epub 2015 Aug 27. Am J Hum Genet. 2015. PMID: 26320891 Free PMC article.
-
Massive sequencing of 70 genes reveals a myriad of missing genes or mechanisms to be uncovered in hereditary spastic paraplegias.Eur J Hum Genet. 2017 Nov;25(11):1217-1228. doi: 10.1038/ejhg.2017.124. Epub 2017 Aug 23. Eur J Hum Genet. 2017. PMID: 28832565 Free PMC article.
-
Genetic diagnosis of Mendelian disorders via RNA sequencing.Nat Commun. 2017 Jun 12;8:15824. doi: 10.1038/ncomms15824. Nat Commun. 2017. PMID: 28604674 Free PMC article.
References
-
- Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin–rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 2002; 30: 97–101. - PubMed
-
- Baumgartner MR, Hu CA, Almashanu S, Steel G, Obie C, Aral B, et al. Hyperammonemia with reduced ornithine, citrulline, arginine and proline: a new inborn error caused by a mutation in the gene encoding delta(1)-pyrroline-5-carboxylate synthase. Hum Mol Genet 2000; 9: 2853–8. - PubMed
-
- Baumgartner MR, Rabier D, Nassogne MC, Dufier JL, Padovani JP, Kamoun P, et al. Delta1-pyrroline-5-carboxylate synthase deficiency: neurodegeneration, cataracts and connective tissue manifestations combined with hyperammonaemia and reduced ornithine, citrulline, arginine and proline. Eur J Pediatr 2005; 164: 31–6. - PubMed
-
- Bicknell LS, Pitt J, Aftimos S, Ramadas R, Maw MA, Robertson SP. A missense mutation in ALDH18A1, encoding Delta1-pyrroline-5-carboxylate synthase (P5CS), causes an autosomal recessive neurocutaneous syndrome. Eur J Hum Genet 2008; 16: 1176–86. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
