

Targeting EZH2 and PRC2 dependence as novel anticancer therapy
- PMID: 26027790
- PMCID: PMC4706459
- DOI: 10.1016/j.exphem.2015.05.001
Targeting EZH2 and PRC2 dependence as novel anticancer therapy
Abstract
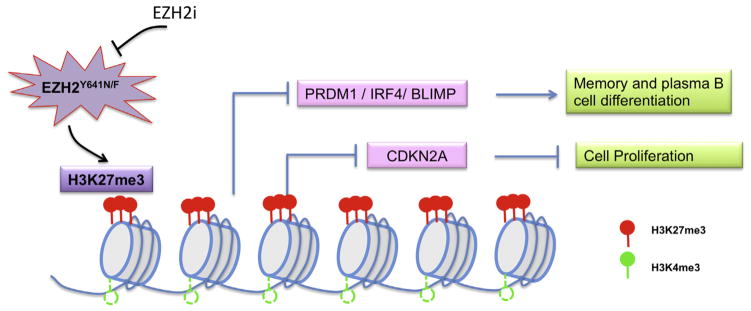
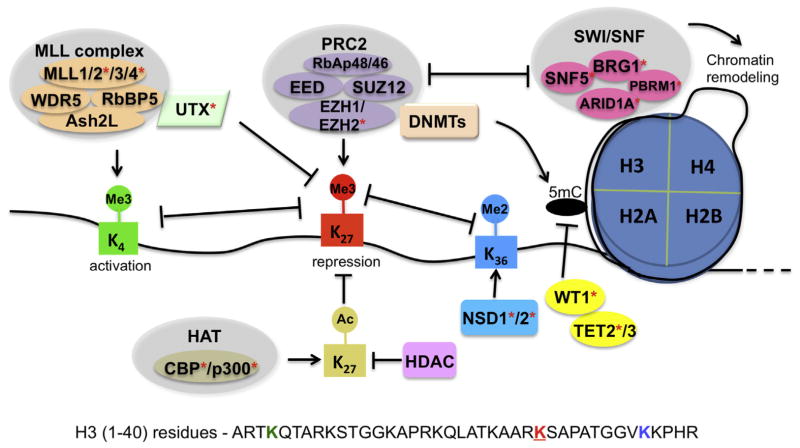
Distinctive patterns of chromatin modification control gene expression and define cellular identity during development and cell differentiation. Polycomb repressive complex 2 (PRC2), the sole mammalian enzymatic complex capable of establishing gene-repressive high-degree methylation of histone H3 at lysine 27 (H3K27), plays crucial roles in regulation of normal and malignant hematopoiesis. Recently, increasing evidence has indicated that recurrent gain-of-function mutation and overexpression of EZH2, the catalytic subunit of PRC2, drive and promote malignant transformation such as B-cell lymphomagenesis, providing a rationale for PRC2 inhibition as a novel anticancer strategy. Here, we summarize the recently developed strategies for inhibition of PRC2, which include a series of highly specific, highly potent, small-molecule inhibitors of EZH2 and EZH1, an EZH2-related methyltransferase. PRC2 establishes functional crosstalk with numerous epigenetic machineries during dynamic regulation of gene transcription. Perturbation of such functional crosstalk caused by genetic events observed in various hematologic cancers, such as inactivation of SNF5 and somatic mutation of UTX, confers PRC2 dependence, thus rendering an increased sensitivity to PRC2 inhibition. We discuss our current understanding of EZH2 somatic mutations frequently found in B-cell lymphomas and recurrent mutations in various other epigenetic regulators as novel molecular predictors and determinants of PRC2 sensitivity. As recent advances have indicated a critical developmental or tumor-suppressive role for PRC2 and EZH2 in various tissue types, we discuss concerns over potentially toxic or even adverse effects associated with EZH2/1 inhibition in certain biological contexts or on cancer genetic background. Collectively, inhibition of PRC2 catalytic activity has emerged as a promising therapeutic intervention for the precise treatment of a range of genetically defined hematologic malignancies and can be potentially applied to a broader spectrum of human cancers that bear similar genetic and epigenetic characteristics.
Published by Elsevier Inc.
Conflict of interest statement
The authors have no conflicting financial interests to disclose.
Figures



References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
