

Redox effects and cytotoxic profiles of MJ25 and auranofin towards malignant melanoma cells
- PMID: 26029997
- PMCID: PMC4599284
- DOI: 10.18632/oncotarget.4108
Redox effects and cytotoxic profiles of MJ25 and auranofin towards malignant melanoma cells
Abstract
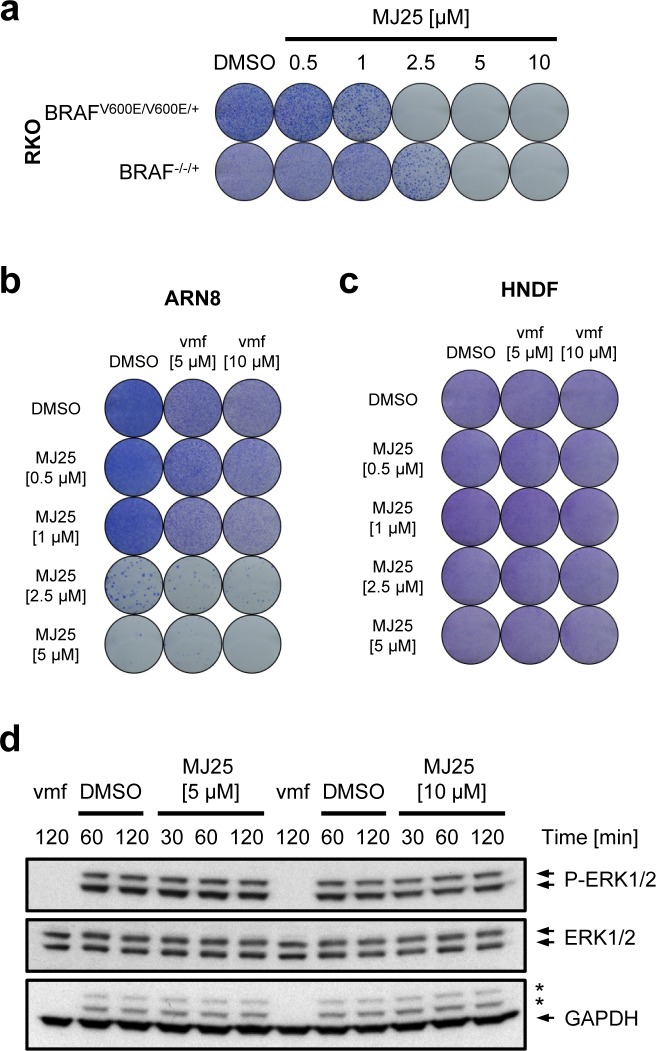
Malignant melanoma is the most dangerous type of skin cancer. Although recent progress in treatment has been achieved, lack of response, drug resistance and relapse remain major problems. The tumor suppressor p53 is rarely mutated in melanoma, yet it is inactive in the majority of cases due to dysregulation of upstream pathways. Thus, we screened for compounds that can activate p53 in melanoma cells. Here we describe effects of the small molecule MJ25 (2-{[2-(1,3-benzothiazol-2-ylsulfonyl)ethyl]thio}-1,3-benzoxazole), which increased the level of p53-dependent transactivation both as a single agent and in combination with nutlin-3. Furthermore, MJ25 showed potent cytotoxicity towards melanoma cell lines, whilst having weaker effects against human normal cells. MJ25 was also identified in an independent screen as an inhibitor of thioredoxin reductase 1 (TrxR1), an important selenoenzyme in the control of oxidative stress and redox regulation. The well-characterized TrxR inhibitor auranofin, which is FDA-approved and currently in clinical trials against leukemia and a number of solid cancers, displayed effects comparable with MJ25 on cells and led to eradication of cultured melanoma cells at low micromolar concentrations. In conclusion, auranofin, MJ25 or other inhibitors of TrxR1 should be evaluated as candidate compounds or leads for targeted therapy of malignant melanoma.
Keywords: auranofin; malignant melanoma; p53; thioredoxin reductase 1; vemurafenib.
Conflict of interest statement
The authors declare no conflict of interest.
Figures






References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
