

Additive regulation of adiponectin expression by the mediterranean diet olive oil components oleic Acid and hydroxytyrosol in human adipocytes
- PMID: 26030149
- PMCID: PMC4452359
- DOI: 10.1371/journal.pone.0128218
Additive regulation of adiponectin expression by the mediterranean diet olive oil components oleic Acid and hydroxytyrosol in human adipocytes
Abstract
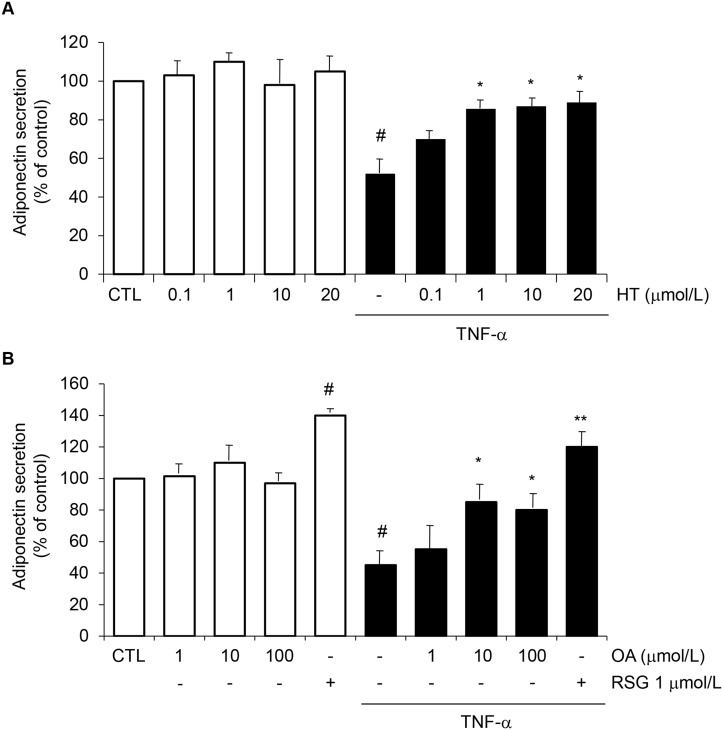
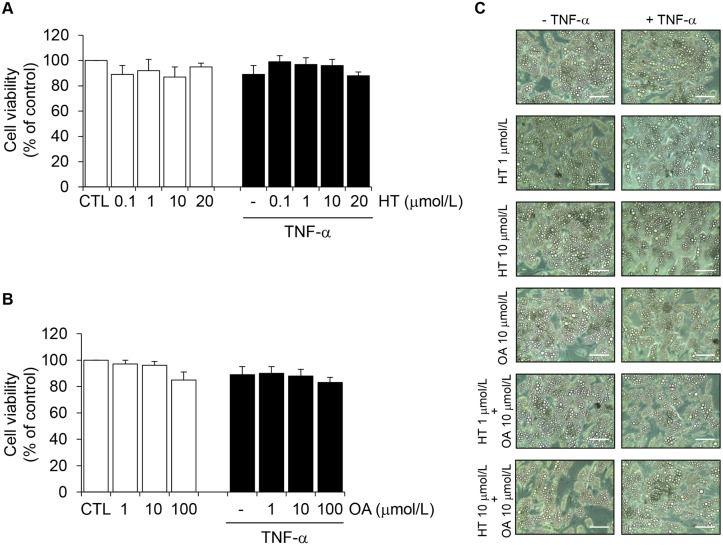
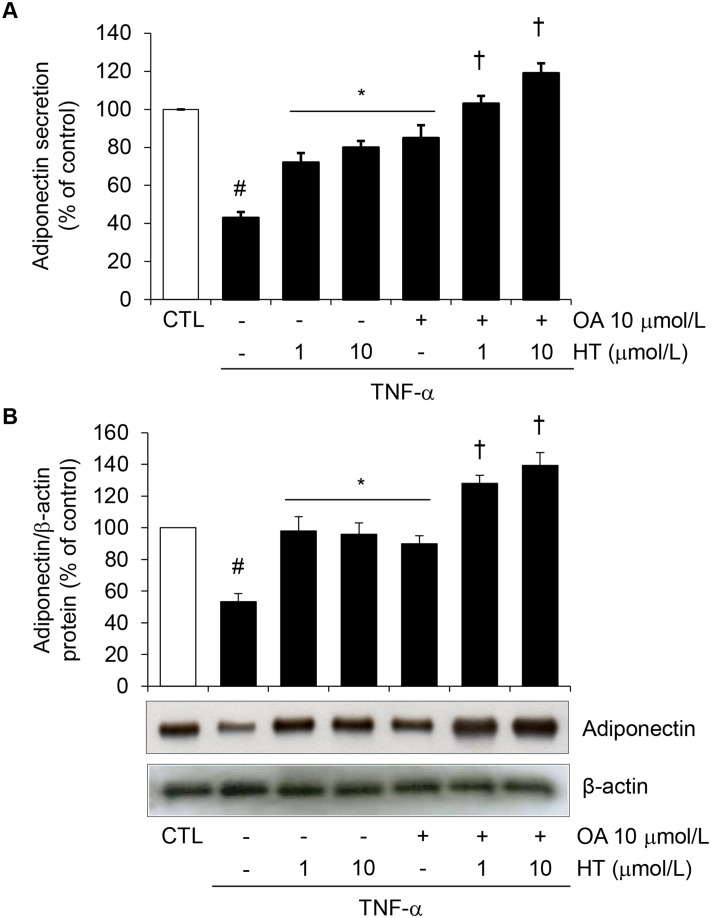
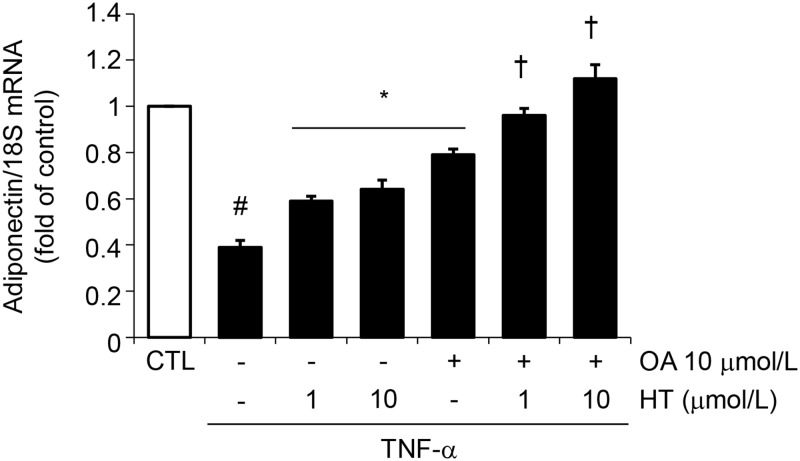
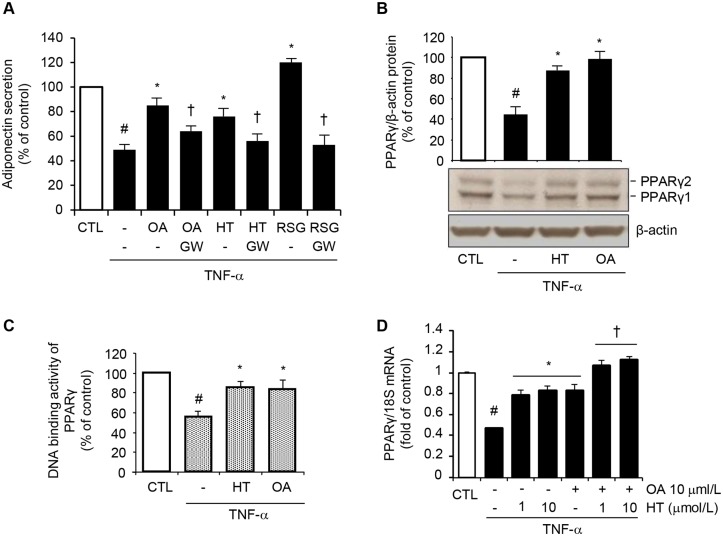
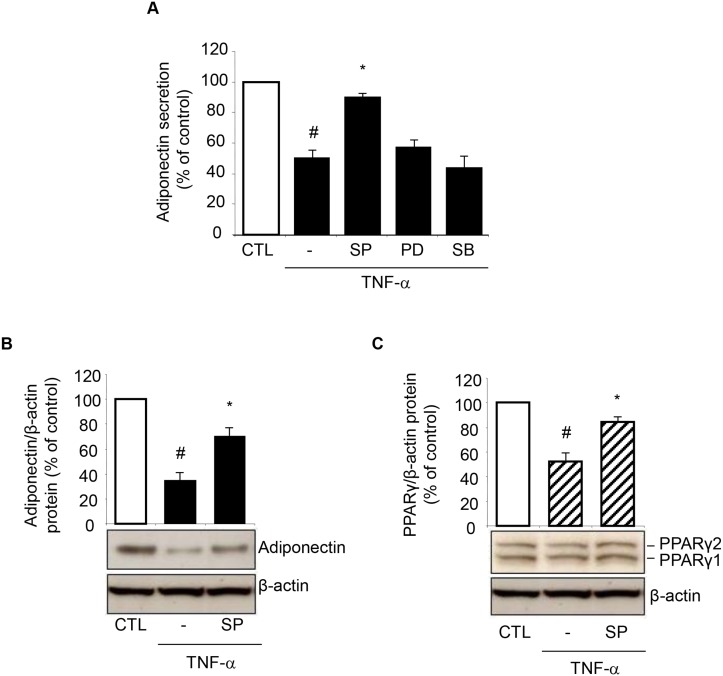
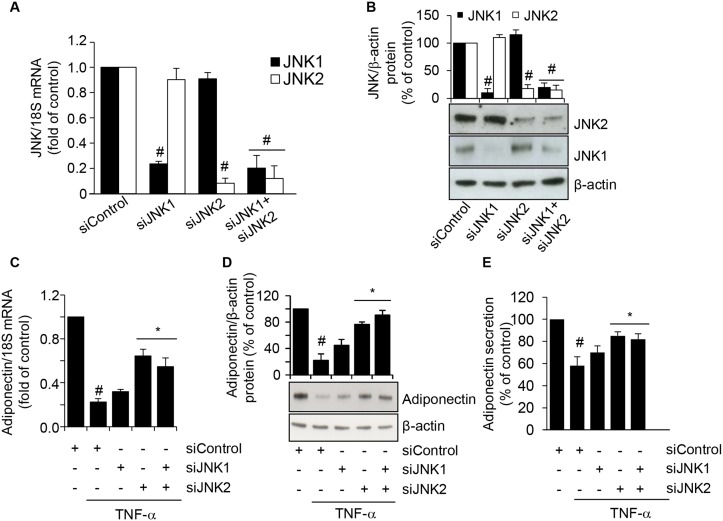
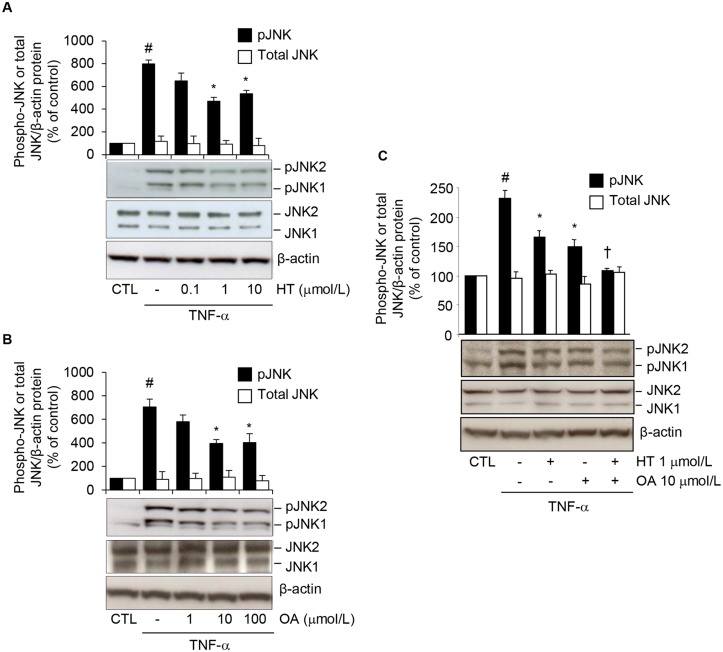
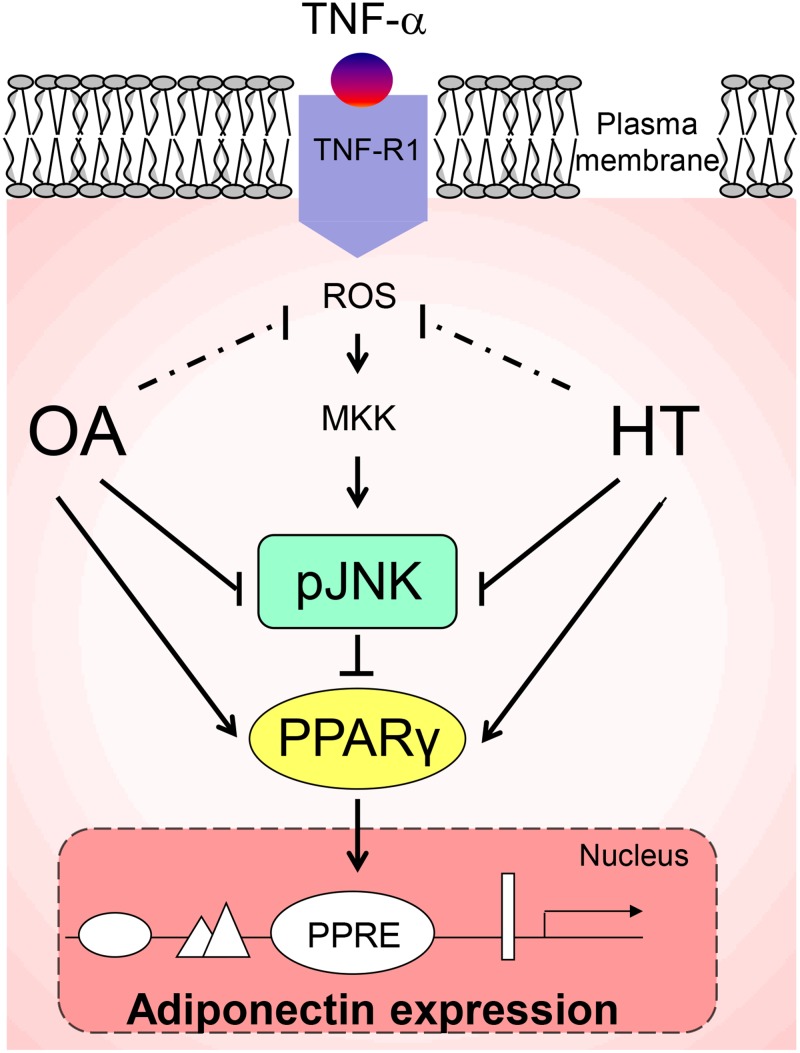
Adiponectin, an adipocyte-derived insulin-sensitizing and anti-inflammatory hormone, is suppressed in obesity through mechanisms involving chronic inflammation and oxidative stress. Olive oil consumption is associated with beneficial cardiometabolic actions, with possible contributions from the antioxidant phenol hydroxytyrosol (HT) and the monounsaturated fatty acid oleic acid (OA, 18:1n-9 cis), both possessing anti-inflammatory and vasculo-protective properties. We determined the effects of HT and OA, alone and in combination, on adiponectin expression in human and murine adipocytes under pro-inflammatory conditions induced by the cytokine tumor necrosis factor(TNF)-α. We used human Simpson-Golabi-Behmel syndrome (SGBS) adipocytes and murine 3T3-L1 adipocytes as cell model systems, and pretreated them with 1-100 μmol/L OA, 0.1-20 μmol/L HT or OA plus HT combination before stimulation with 10 ng/mL TNF-α. OA or HT significantly (P<0.05) prevented TNF-α-induced suppression of total adiponectin secretion (by 42% compared with TNF-α alone) as well as mRNA levels (by 30% compared with TNF-α alone). HT and OA also prevented-by 35%-TNF-α-induced downregulation of peroxisome proliferator-activated receptor PPARγ. Co-treatment with HT and OA restored adiponectin and PPARγ expression in an additive manner compared with single treatments. Exploring the activation of JNK, which is crucial for both adiponectin and PPARγ suppression by TNF-α, we found that HT and OA additively attenuated TNF-α-stimulated JNK phosphorylation (up to 55% inhibition). In conclusion, the virgin olive oil components OA and HT, at nutritionally relevant concentrations, have additive effects in preventing adiponectin downregulation in inflamed adipocytes through an attenuation of JNK-mediated PPARγ suppression.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
