

A metagenomic survey of viral abundance and diversity in mosquitoes from Hubei province
- PMID: 26030271
- PMCID: PMC4452694
- DOI: 10.1371/journal.pone.0129845
A metagenomic survey of viral abundance and diversity in mosquitoes from Hubei province
Abstract
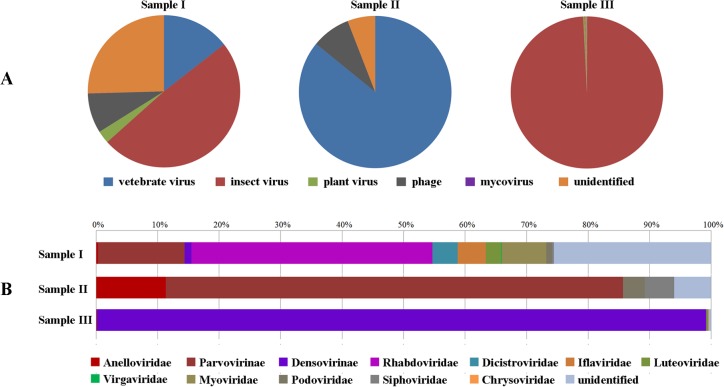
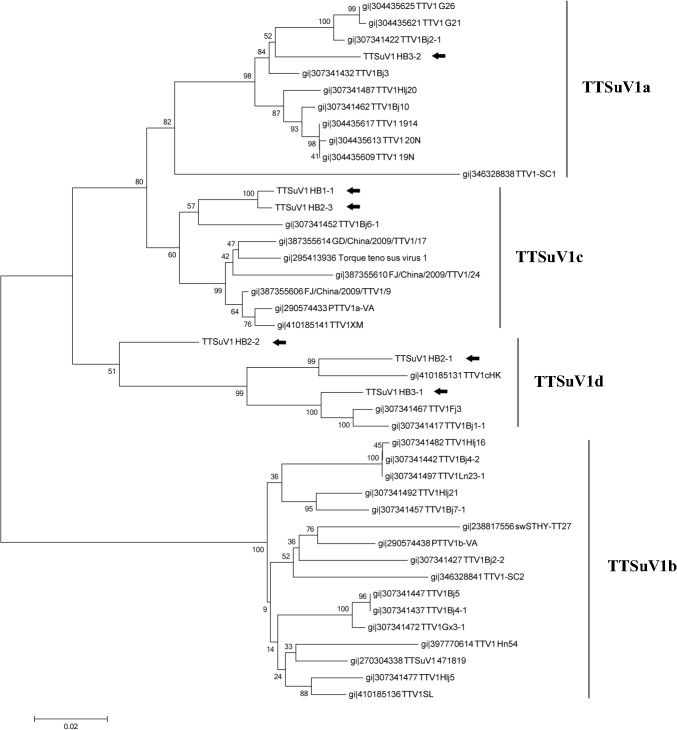
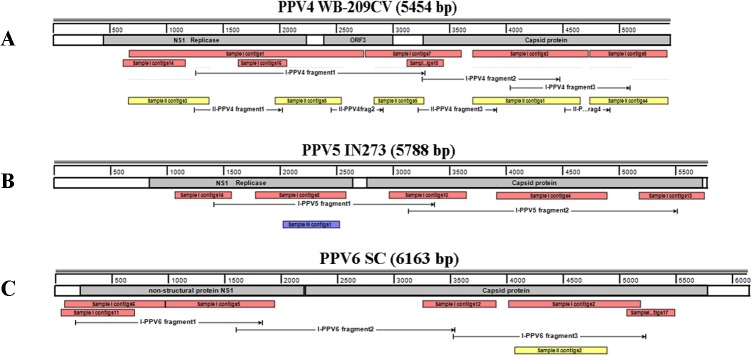
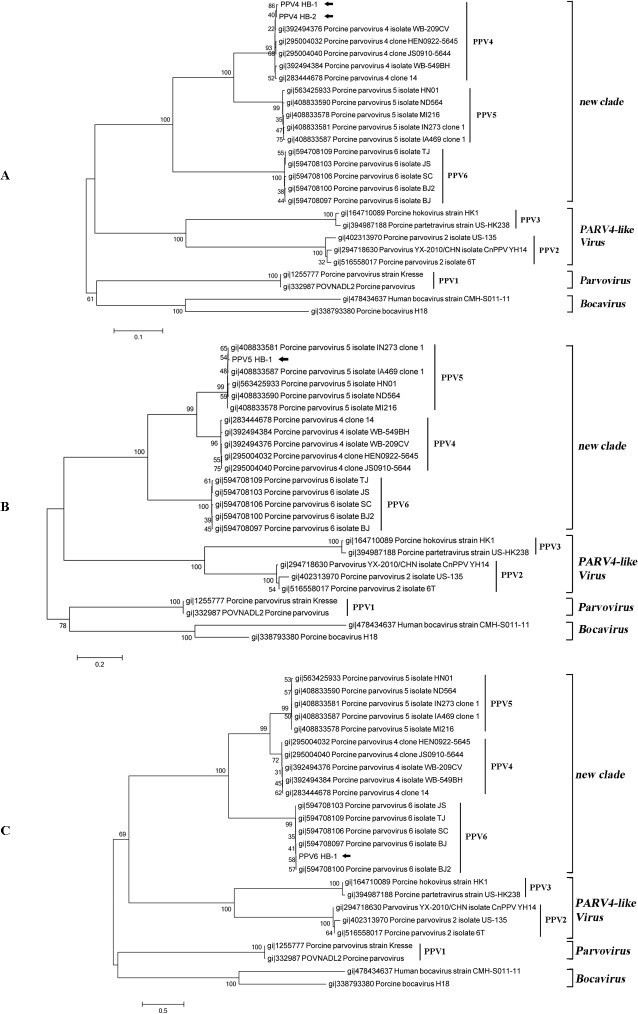
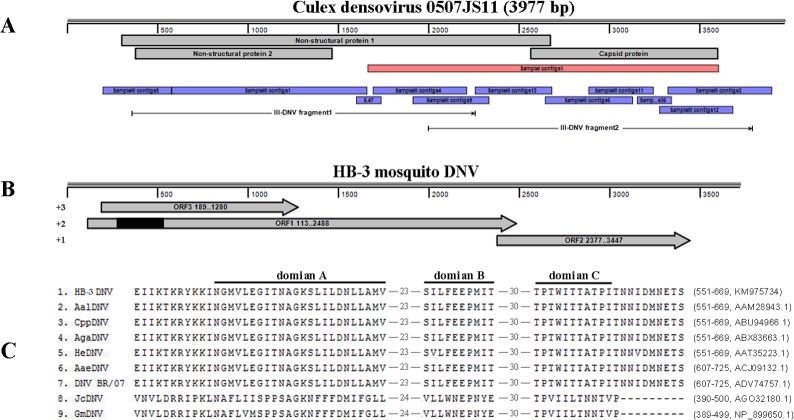
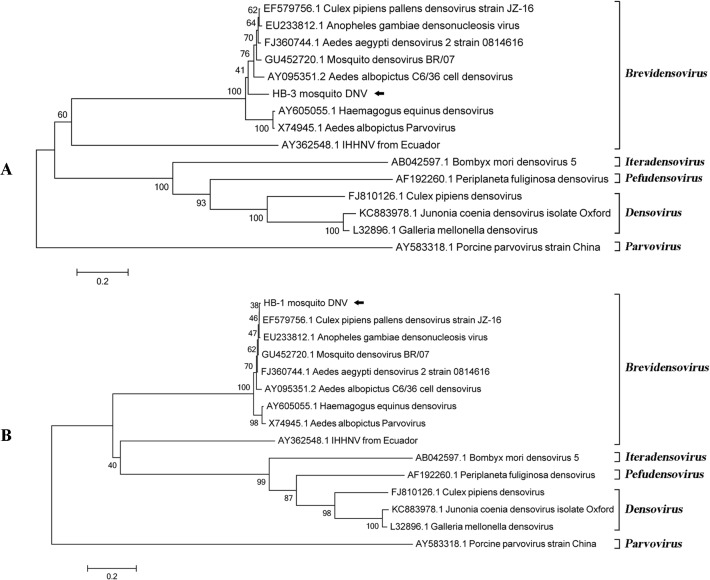
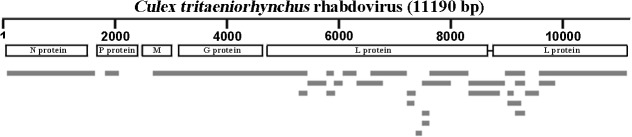
Mosquitoes as one of the most common but important vectors have the potential to transmit or acquire a lot of viruses through biting, however viral flora in mosquitoes and its impact on mosquito-borne disease transmission has not been well investigated and evaluated. In this study, the metagenomic techniquehas been successfully employed in analyzing the abundance and diversity of viral community in three mosquito samples from Hubei, China. Among 92,304 reads produced through a run with 454 GS FLX system, 39% have high similarities with viral sequences belonging to identified bacterial, fungal, animal, plant and insect viruses, and 0.02% were classed into unidentified viral sequences, demonstrating high abundance and diversity of viruses in mosquitoes. Furthermore, two novel viruses in subfamily Densovirinae and family Dicistroviridae were identified, and six torque tenosus virus1 in family Anelloviridae, three porcine parvoviruses in subfamily Parvovirinae and a Culex tritaeniorhynchus rhabdovirus in Family Rhabdoviridae were preliminarily characterized. The viral metagenomic analysis offered us a deep insight into the viral population of mosquito which played an important role in viral initiative or passive transmission and evolution during the process.
Conflict of interest statement
Figures

References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
