

Cardiac metabolic pathways affected in the mouse model of barth syndrome
- PMID: 26030409
- PMCID: PMC4451073
- DOI: 10.1371/journal.pone.0128561
Cardiac metabolic pathways affected in the mouse model of barth syndrome
Abstract
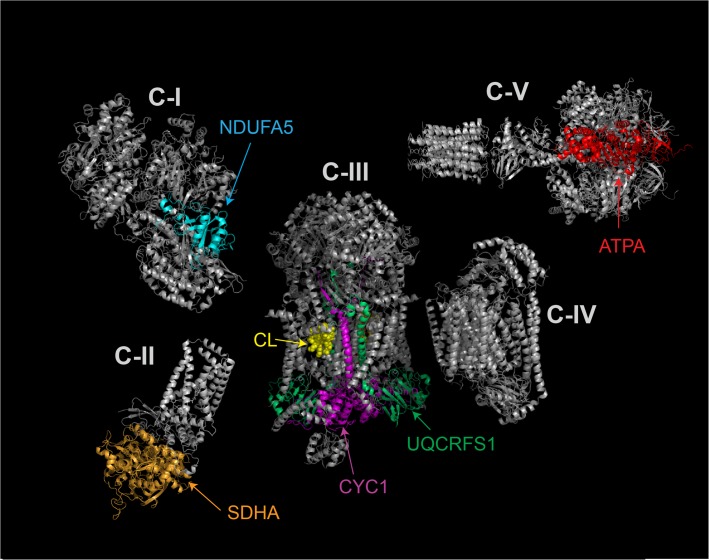
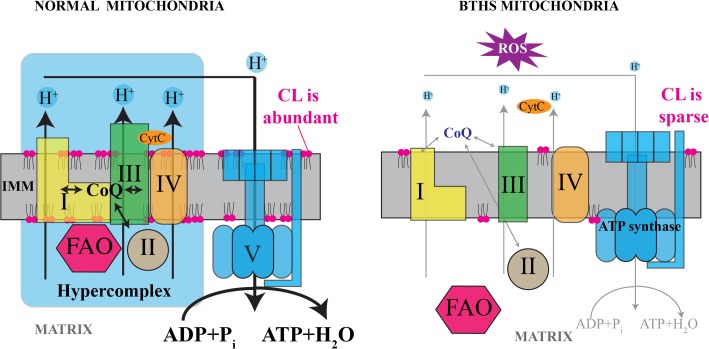
Cardiolipin (CL) is a mitochondrial phospholipid essential for electron transport chain (ETC) integrity. CL-deficiency in humans is caused by mutations in the tafazzin (Taz) gene and results in a multisystem pediatric disorder, Barth syndrome (BTHS). It has been reported that tafazzin deficiency destabilizes mitochondrial respiratory chain complexes and affects supercomplex assembly. The aim of this study was to investigate the impact of Taz-knockdown on the mitochondrial proteomic landscape and metabolic processes, such as stability of respiratory chain supercomplexes and their interactions with fatty acid oxidation enzymes in cardiac muscle. Proteomic analysis demonstrated reduction of several polypeptides of the mitochondrial respiratory chain, including Rieske and cytochrome c1 subunits of complex III, NADH dehydrogenase alpha subunit 5 of complex I and the catalytic core-forming subunit of F0F1-ATP synthase. Taz gene knockdown resulted in upregulation of enzymes of folate and amino acid metabolic pathways in heart mitochondria, demonstrating that Taz-deficiency causes substantive metabolic remodeling in cardiac muscle. Mitochondrial respiratory chain supercomplexes are destabilized in CL-depleted mitochondria from Taz knockdown hearts resulting in disruption of the interactions between ETC and the fatty acid oxidation enzymes, very long-chain acyl-CoA dehydrogenase and long-chain 3-hydroxyacyl-CoA dehydrogenase, potentially affecting the metabolic channeling of reducing equivalents between these two metabolic pathways. Mitochondria-bound myoglobin was significantly reduced in Taz-knockdown hearts, potentially disrupting intracellular oxygen delivery to the oxidative phosphorylation system. Our results identify the critical pathways affected by the Taz-deficiency in mitochondria and establish a future framework for development of therapeutic options for BTHS.
Conflict of interest statement
Figures
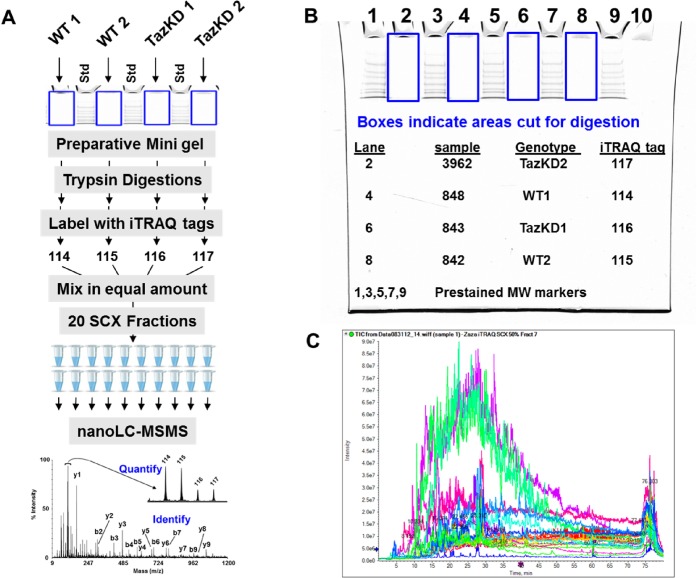
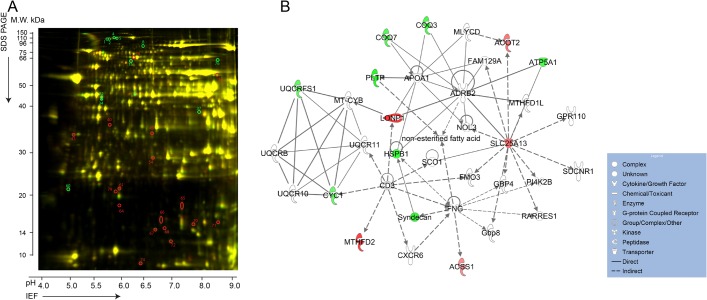
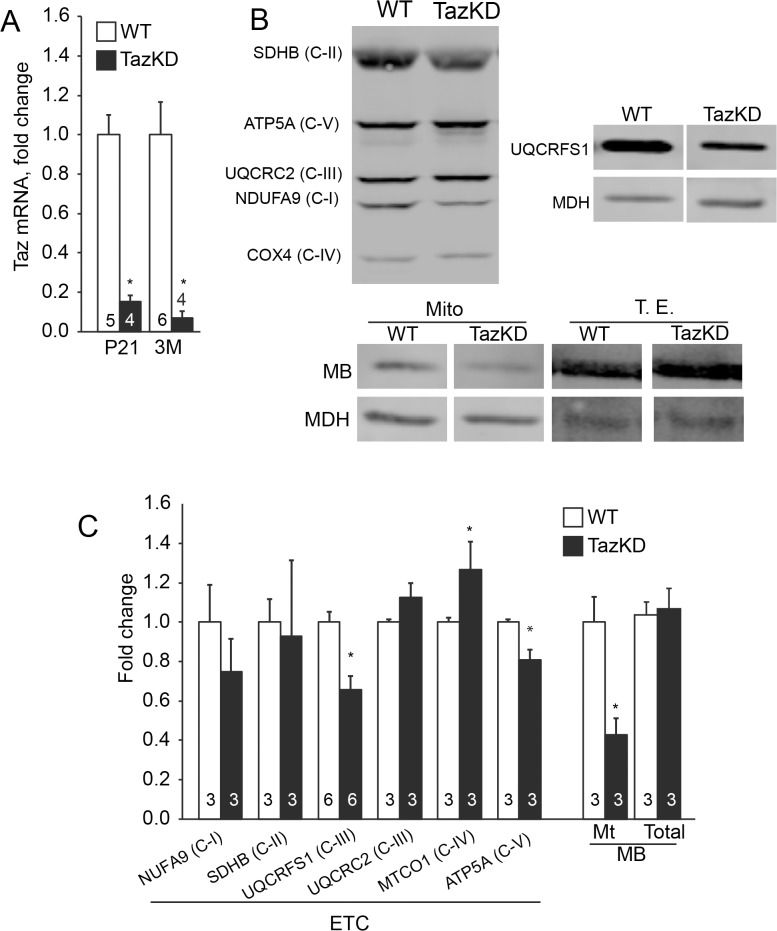

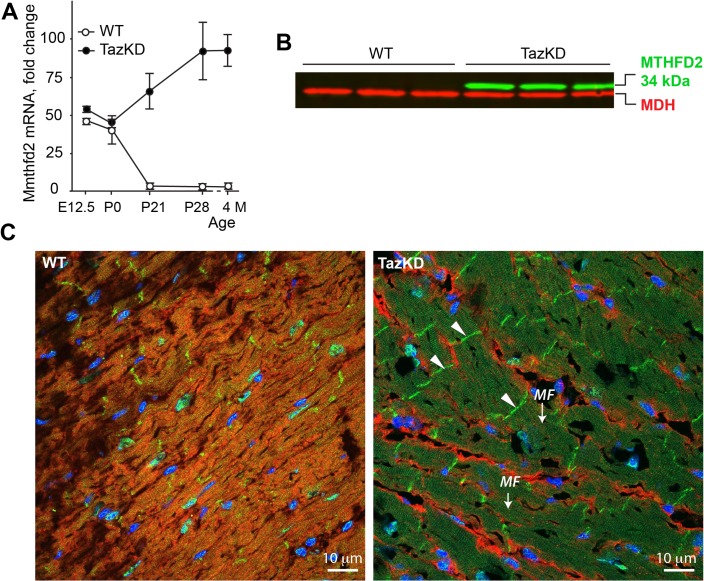
References
-
- Schlame M, Ren M, Xu Y, Greenberg ML, Haller I. Molecular symmetry in mitochondrial cardiolipins. Chemistry and physics of lipids. 2005;138(1–2):38–49. . - PubMed
-
- Chicco AJ, Sparagna GC. Role of cardiolipin alterations in mitochondrial dysfunction and disease. Am J Physiol Cell Physiol. 2007;292(1):C33–44. . - PubMed
-
- Vreken P, Valianpour F, Nijtmans LG, Grivell LA, Plecko B, Wanders RJ, et al. Defective remodeling of cardiolipin and phosphatidylglycerol in Barth syndrome. Biochem Biophys Res Commun. 2000;279(2):378–82. . - PubMed
-
- Houtkooper RH, Rodenburg RJ, Thiels C, van Lenthe H, Stet F, Poll-The BT, et al. Cardiolipin and monolysocardiolipin analysis in fibroblasts, lymphocytes, and tissues using high-performance liquid chromatography-mass spectrometry as a diagnostic test for Barth syndrome. Anal Biochem. 2009;387(2):230–7. Epub 2009/05/21. doi: S0003-2697(09)00060-8 [pii] 10.1016/j.ab.2009.01.032 . - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
