

Immune cell profiling to guide therapeutic decisions in rheumatic diseases
- PMID: 26034835
- PMCID: PMC4898649
- DOI: 10.1038/nrrheum.2015.71
Immune cell profiling to guide therapeutic decisions in rheumatic diseases
Abstract
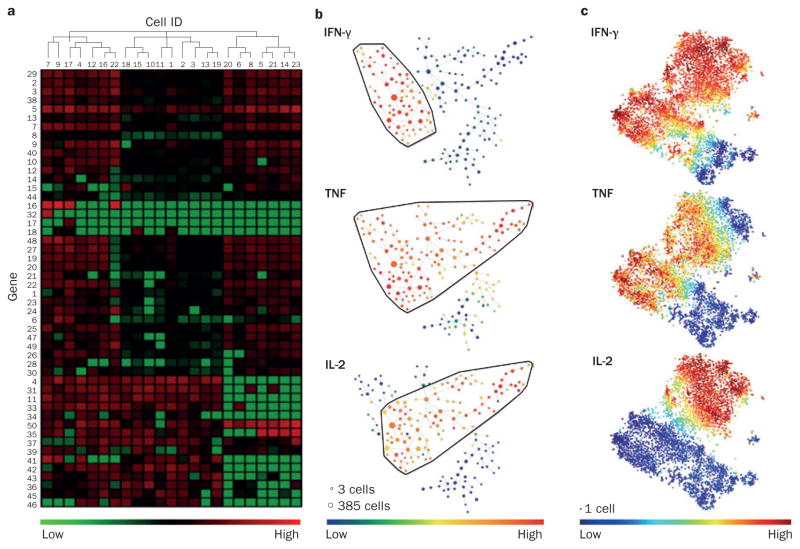
Biomarkers are needed to guide treatment decisions for patients with rheumatic diseases. Although the phenotypic and functional analysis of immune cells is an appealing strategy for understanding immune-mediated disease processes, immune cell profiling currently has no role in clinical rheumatology. New technologies, including mass cytometry, gene expression profiling by RNA sequencing (RNA-seq) and multiplexed functional assays, enable the analysis of immune cell function with unprecedented detail and promise not only a deeper understanding of pathogenesis, but also the discovery of novel biomarkers. The large and complex data sets generated by these technologies--big data--require specialized approaches for analysis and visualization of results. Standardization of assays and definition of the range of normal values are additional challenges when translating these novel approaches into clinical practice. In this Review, we discuss technological advances in the high-dimensional analysis of immune cells and consider how these developments might support the discovery of predictive biomarkers to benefit the practice of rheumatology and improve patient care.
Conflict of interest statement
The authors declare no competing interests.
Figures




References
-
- Emery P, Dörner T. Optimising treatment in rheumatoid arthritis: a review of potential biological markers of response. Ann Rheum Dis. 2011;70:2063–2070. - PubMed
-
- Virgo PF, Gibbs GJ. Flow cytometry in clinical pathology. Ann Clin Biochem. 2012;49:17–28. - PubMed
-
- Kittleson MM, Kobashigawa JA. Long-term care of the heart transplant recipient. Curr Opin Organ Transplant. 2014;19:515–524. - PubMed
-
- Mirnezami R, Nicholson J, Darzi A. Preparing for precision medicine. N Engl J Med. 2012;366:489–491. - PubMed
Publication types
MeSH terms
Grants and funding
- R03AR066357‑01A1/AR/NIAMS NIH HHS/United States
- T32 5T32AR007530/AR/NIAMS NIH HHS/United States
- 1R01AR063759-01A1/AR/NIAMS NIH HHS/United States
- 1U01HG0070033/HG/NHGRI NIH HHS/United States
- 5U01GM092691-04/GM/NIGMS NIH HHS/United States
- UH2 AR067677/AR/NIAMS NIH HHS/United States
- 1R01AR065183-01/AR/NIAMS NIH HHS/United States
- 1UH2AR067694‑01/AR/NIAMS NIH HHS/United States
- 1UH2AR067677-01/AR/NIAMS NIH HHS/United States
- R03 AR066357/AR/NIAMS NIH HHS/United States
- U19 AI111224/AI/NIAID NIH HHS/United States
- T32 AR007530/AR/NIAMS NIH HHS/United States
- U01 GM092691/GM/NIGMS NIH HHS/United States
- R01 AR063759/AR/NIAMS NIH HHS/United States
- R01 AR062886/AR/NIAMS NIH HHS/United States
- R01 AR065183/AR/NIAMS NIH HHS/United States
- UH2 AR067694/AR/NIAMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
