

The Meckel-Gruber syndrome protein TMEM67 controls basal body positioning and epithelial branching morphogenesis in mice via the non-canonical Wnt pathway
- PMID: 26035863
- PMCID: PMC4457033
- DOI: 10.1242/dmm.019083
The Meckel-Gruber syndrome protein TMEM67 controls basal body positioning and epithelial branching morphogenesis in mice via the non-canonical Wnt pathway
Abstract
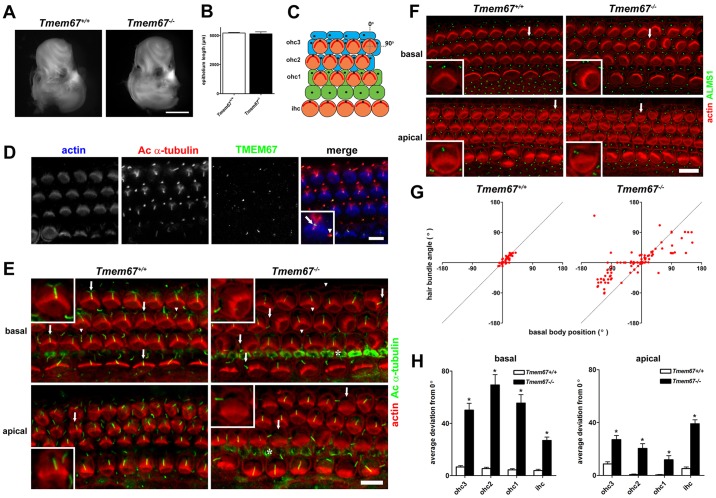
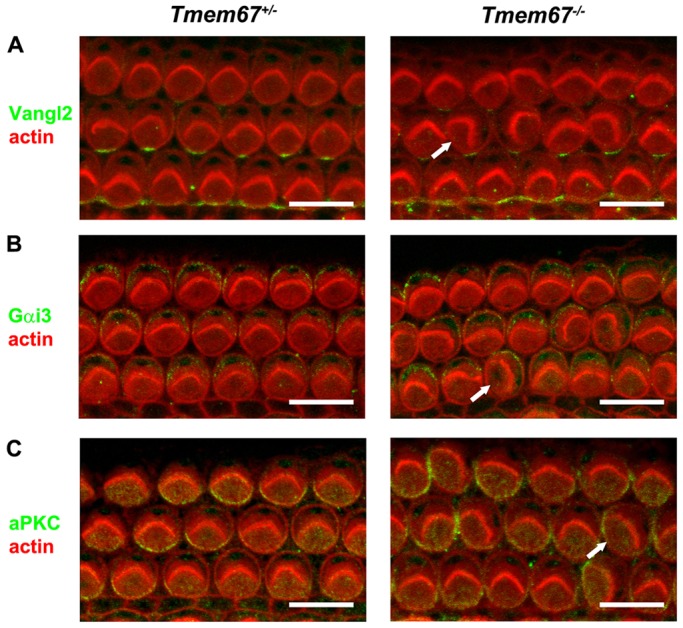
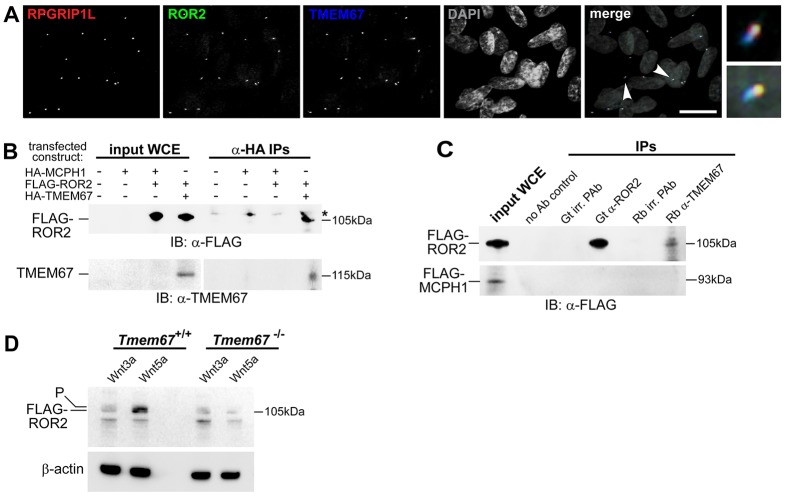
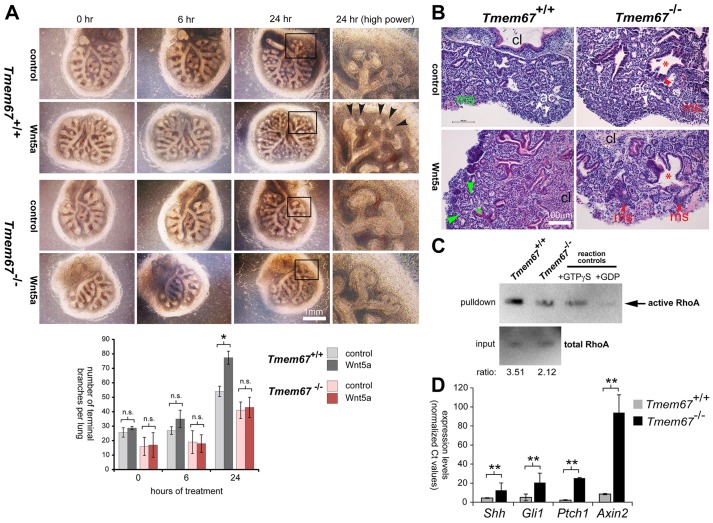
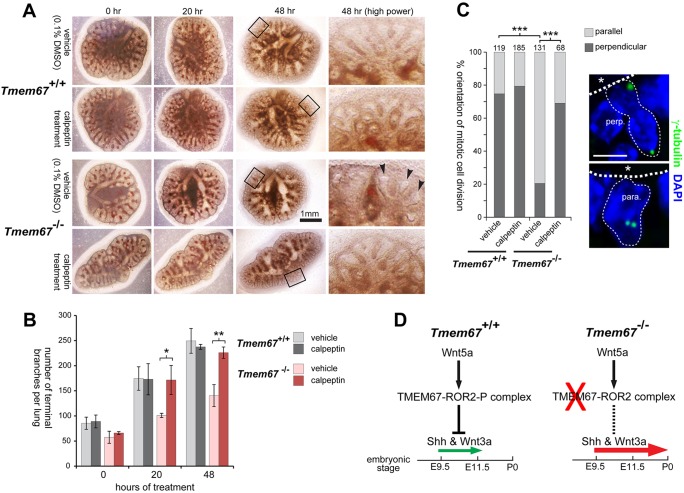
Ciliopathies are a group of developmental disorders that manifest with multi-organ anomalies. Mutations in TMEM67 (MKS3) cause a range of human ciliopathies, including Meckel-Gruber and Joubert syndromes. In this study we describe multi-organ developmental abnormalities in the Tmem67(tm1Dgen/H1) knockout mouse that closely resemble those seen in Wnt5a and Ror2 knockout mice. These include pulmonary hypoplasia, ventricular septal defects, shortening of the body longitudinal axis, limb abnormalities, and cochlear hair cell stereociliary bundle orientation and basal body/kinocilium positioning defects. The basal body/kinocilium complex was often uncoupled from the hair bundle, suggesting aberrant basal body migration, although planar cell polarity and apical planar asymmetry in the organ of Corti were normal. TMEM67 (meckelin) is essential for phosphorylation of the non-canonical Wnt receptor ROR2 (receptor-tyrosine-kinase-like orphan receptor 2) upon stimulation with Wnt5a-conditioned medium. ROR2 also colocalises and interacts with TMEM67 at the ciliary transition zone. Additionally, the extracellular N-terminal domain of TMEM67 preferentially binds to Wnt5a in an in vitro binding assay. Cultured lungs of Tmem67 mutant mice failed to respond to stimulation of epithelial branching morphogenesis by Wnt5a. Wnt5a also inhibited both the Shh and canonical Wnt/β-catenin signalling pathways in wild-type embryonic lung. Pulmonary hypoplasia phenotypes, including loss of correct epithelial branching morphogenesis and cell polarity, were rescued by stimulating the non-canonical Wnt pathway downstream of the Wnt5a-TMEM67-ROR2 axis by activating RhoA. We propose that TMEM67 is a receptor that has a main role in non-canonical Wnt signalling, mediated by Wnt5a and ROR2, and normally represses Shh signalling. Downstream therapeutic targeting of the Wnt5a-TMEM67-ROR2 axis might, therefore, reduce or prevent pulmonary hypoplasia in ciliopathies and other congenital conditions.
Keywords: Ciliopathy; Hair bundle; Kinocilia; MKS3; Meckelin; PCP; Planar cell polarity; Primary cilia; Stereocilia; TMEM67; Wnt signalling.
© 2015. Published by The Company of Biologists Ltd.
Conflict of interest statement
The authors declare no competing or financial interests.
Figures


References
-
- Abdelhamed Z. A., Wheway G., Szymanska K., Natarajan S., Toomes C., Inglehearn C. and Johnson C. A. (2013). Variable expressivity of ciliopathy neurological phenotypes that encompass Meckel–Gruber syndrome and Joubert syndrome is caused by complex de-regulated ciliogenesis, Shh and Wnt signalling defects. Hum. Mol. Genet. 22, 1358-1372. 10.1093/hmg/dds546 - DOI - PMC - PubMed
-
- Arts H. H., Doherty D., van Beersum S. E. C., Parisi M. A., Letteboer S. J. F., Gorden N. T., Peters T. A., Märker T., Voesenek K., Kartono A. et al. (2007). Mutations in the gene encoding the basal body protein RPGRIP1L, a nephrocystin-4 interactor, cause Joubert syndrome. Nat. Genet. 39, 882-888. 10.1038/ng2069 - DOI - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
