

Direct ChIP-Seq significance analysis improves target prediction
- PMID: 26040656
- PMCID: PMC4460594
- DOI: 10.1186/1471-2164-16-S5-S4
Direct ChIP-Seq significance analysis improves target prediction
Abstract
Background: Chromatin immunoprecipitation followed by sequencing of protein-bound DNA fragments (ChIP-Seq) is an effective high-throughput methodology for the identification of context specific DNA fragments that are bound by specific proteins in vivo. Despite significant progress in the bioinformatics analysis of this genome-scale data, a number of challenges remain as technology-dependent biases, including variable target accessibility and mappability, sequence-dependent variability, and non-specific binding affinity must be accounted for.
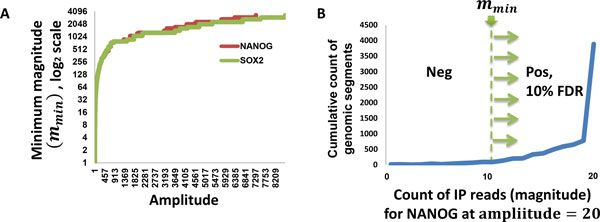
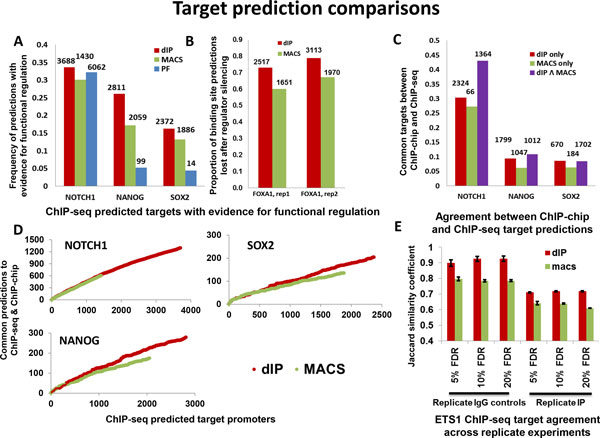
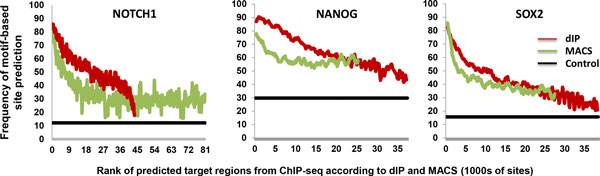
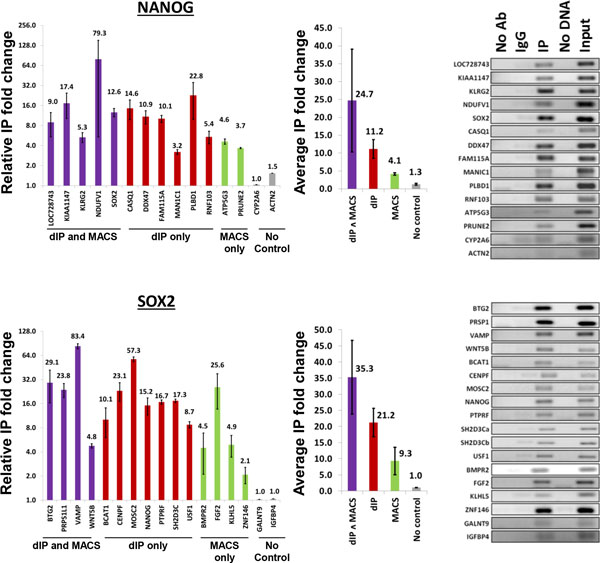
Results and discussion: We introduce a nonparametric method for scoring consensus regions of aligned immunoprecipitated DNA fragments when appropriate control experiments are available. Our method uses local models for null binding; these are necessary because binding prediction scores based on global models alone fail to properly account for specialized features of genomic regions and chance pull downs of specific DNA fragments, thus disproportionally rewarding some genomic regions and decreasing prediction accuracy. We make no assumptions about the structure or amplitude of bound peaks, yet we show that our method outperforms leading methods developed using either global or local null hypothesis models for random binding. We test prediction performance by comparing analyses of ChIP-seq, ChIP-chip, motif-based binding-site prediction, and shRNA assays, showing high reproducibility, binding-site enrichment in predicted target regions, and functional regulation of predicted targets.
Conclusions: Given appropriate controls, a direct nonparametric method for identifying transcription-factor targets from ChIP-Seq assays may lead to both higher sensitivity and higher specificity, and should be preferred or used in conjunction with methods that use parametric models for null binding.
Figures
References
-
- Margolin AA, Palomero T, Sumazin P, Califano A, Ferrando AA, Stolovitzky G. ChIP-on-chip significance analysis reveals large-scale binding and regulation by human transcription factor oncogenes. Proceedings of the National Academy of Sciences. 2009;106(1):244–249. doi: 10.1073/pnas.0806445106. - DOI - PMC - PubMed
-
- Bartlett JM, Stirling D. A short history of the polymerase chain reaction. Methods Mol Biol. 2003;226:3–6. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
