

Role of sodium and calcium dysregulation in tachyarrhythmias in sudden cardiac death
- PMID: 26044250
- PMCID: PMC4458704
- DOI: 10.1161/CIRCRESAHA.116.304678
Role of sodium and calcium dysregulation in tachyarrhythmias in sudden cardiac death
Abstract
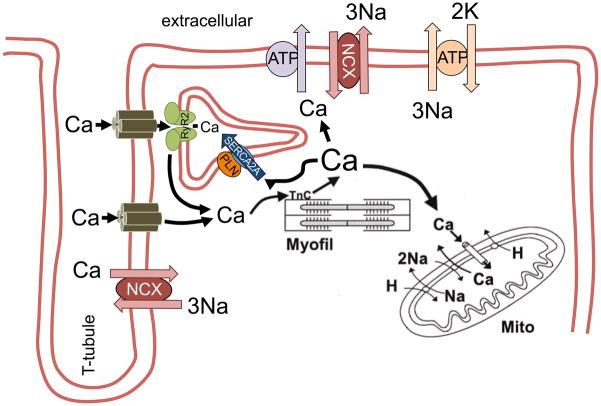
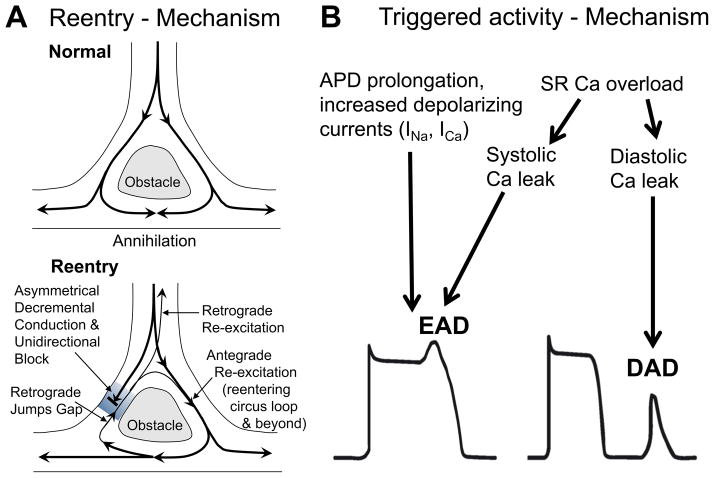
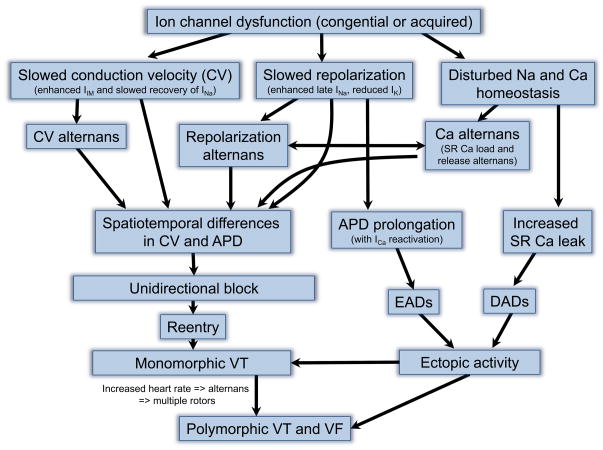
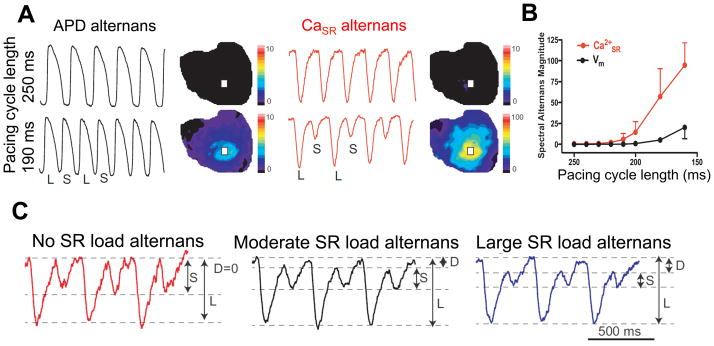
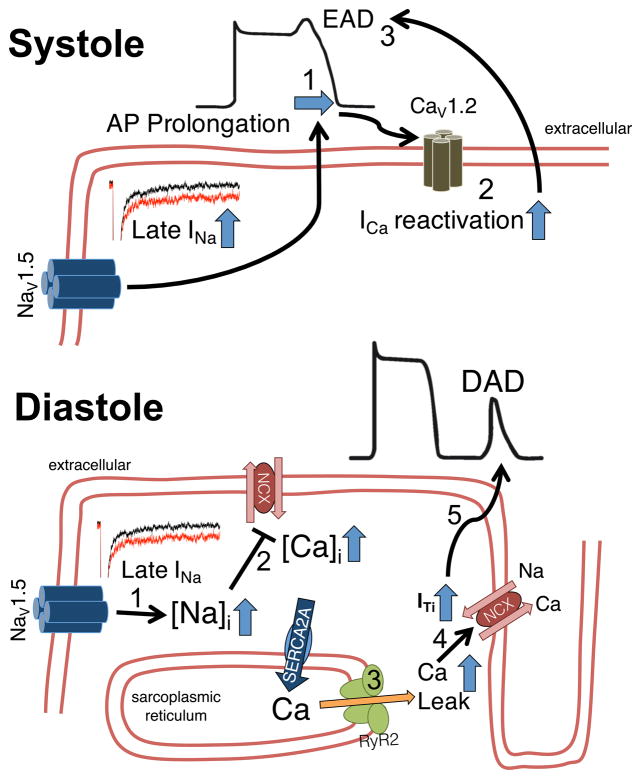
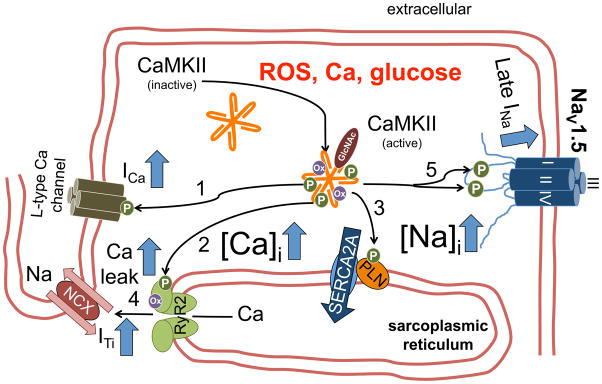
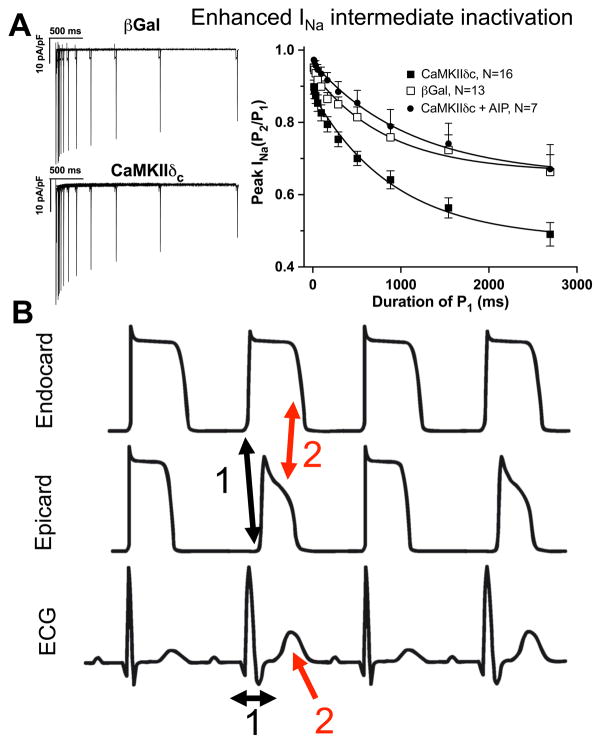
Despite improvements in the therapy of underlying heart disease, sudden cardiac death is a major cause of death worldwide. Disturbed Na and Ca handling is known to be a major predisposing factor for life-threatening tachyarrhythmias. In cardiomyocytes, many ion channels and transporters, including voltage-gated Na and Ca channels, cardiac ryanodine receptors, Na/Ca-exchanger, and SR Ca-ATPase are involved in this regulation. We have learned a lot about the pathophysiological relevance of disturbed ion channel function from monogenetic disorders. Changes in the gating of a single ion channel and the activity of an ion pump suffice to dramatically increase the propensity for arrhythmias even in structurally normal hearts. Nevertheless, patients with heart failure with acquired dysfunction in many ion channels and transporters exhibit profound dysregulation of Na and Ca handling and Ca/calmodulin-dependent protein kinase and are especially prone to arrhythmias. A deeper understanding of the underlying arrhythmic principles is mandatory if we are to improve their outcome. This review addresses basic tachyarrhythmic mechanisms, the underlying ionic mechanisms and the consequences for ion homeostasis, and the situation in complex diseases like heart failure.
Keywords: alternans; arrhythmias; calcium; calcium/calmodulin–dependent kinase II; sodium.
© 2015 American Heart Association, Inc.
Figures
References
-
- Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Judd SE, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Mackey RH, Magid DJ, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, 3rd, Moy CS, Mussolino ME, Neumar RW, Nichol G, Pandey DK, Paynter NP, Reeves MJ, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Heart disease and stroke statistics--2014 update: A report from the american heart association. Circulation. 2014;129:e28–e292. - PMC - PubMed
-
- Lehnart SE, Ackerman MJ, Benson DW, Jr, Brugada R, Clancy CE, Donahue JK, George AL, Jr, Grant AO, Groft SC, January CT, Lathrop DA, Lederer WJ, Makielski JC, Mohler PJ, Moss A, Nerbonne JM, Olson TM, Przywara DA, Towbin JA, Wang LH, Marks AR. Inherited arrhythmias: A national heart, lung, and blood institute and office of rare diseases workshop consensus report about the diagnosis, phenotyping, molecular mechanisms, and therapeutic approaches for primary cardiomyopathies of gene mutations affecting ion channel function. Circulation. 2007;116:2325–2345. - PubMed
-
- Ford ES, Ajani UA, Croft JB, Critchley JA, Labarthe DR, Kottke TE, Giles WH, Capewell S. Explaining the decrease in u.S. Deaths from coronary disease, 1980–2000. N Engl J Med. 2007;356:2388–2398. - PubMed
-
- Dudas K, Lappas G, Stewart S, Rosengren A. Trends in out-of-hospital deaths due to coronary heart disease in sweden (1991 to 2006) Circulation. 2011;123:46–52. - PubMed
-
- Huikuri HV, Castellanos A, Myerburg RJ. Sudden death due to cardiac arrhythmias. N Engl J Med. 2001;345:1473–1482. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
