

Individualized iterative phenotyping for genome-wide analysis of loss-of-function mutations
- PMID: 26046366
- PMCID: PMC4457956
- DOI: 10.1016/j.ajhg.2015.04.013
Individualized iterative phenotyping for genome-wide analysis of loss-of-function mutations
Abstract
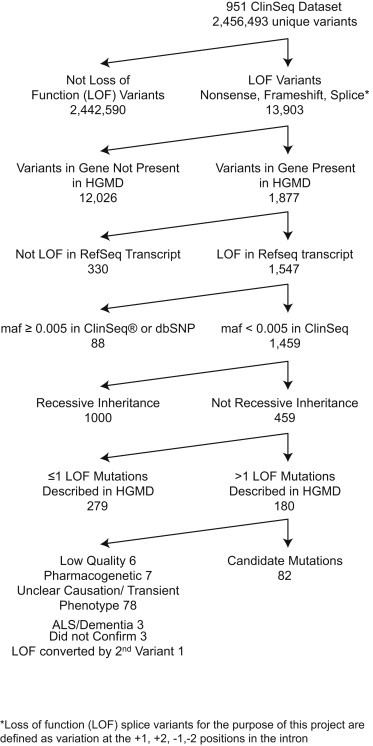
Next-generation sequencing provides the opportunity to practice predictive medicine based on identified variants. Putative loss-of-function (pLOF) variants are common in genomes and understanding their contribution to disease is critical for predictive medicine. To this end, we characterized the consequences of pLOF variants in an exome cohort by iterative phenotyping. Exome data were generated on 951 participants from the ClinSeq cohort and filtered for pLOF variants in genes likely to cause a phenotype in heterozygotes. 103 of 951 exomes had such a pLOF variant and 79 participants were evaluated. Of those 79, 34 had findings or family histories that could be attributed to the variant (28 variants in 18 genes), 2 had indeterminate findings (2 variants in 2 genes), and 43 had no findings or a negative family history for the trait (34 variants in 28 genes). The presence of a phenotype was correlated with two mutation attributes: prior report of pathogenicity for the variant (p = 0.0001) and prior report of other mutations in the same exon (p = 0.0001). We conclude that 1/30 unselected individuals harbor a pLOF mutation associated with a phenotype either in themselves or their family. This is more common than has been assumed and has implications for the setting of prior probabilities of affection status for predictive medicine.
Copyright © 2015 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Xue Y., Chen Y., Ayub Q., Huang N., Ball E.V., Mort M., Phillips A.D., Shaw K., Stenson P.D., Cooper D.N., Tyler-Smith C., 1000 Genomes Project Consortium Deleterious- and disease-allele prevalence in healthy individuals: insights from current predictions, mutation databases, and population-scale resequencing. Am. J. Hum. Genet. 2012;91:1022–1032. - PMC - PubMed
-
- Biesecker L.G., Mullikin J.C., Facio F.M., Turner C., Cherukuri P.F., Blakesley R.W., Bouffard G.G., Chines P.S., Cruz P., Hansen N.F., NISC Comparative Sequencing Program The ClinSeq Project: piloting large-scale genome sequencing for research in genomic medicine. Genome Res. 2009;19:1665–1674. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
