

Differential connectivity of splicing activators and repressors to the human spliceosome
- PMID: 26047612
- PMCID: PMC4502471
- DOI: 10.1186/s13059-015-0682-5
Differential connectivity of splicing activators and repressors to the human spliceosome
Abstract
Background: During spliceosome assembly, protein-protein interactions (PPI) are sequentially formed and disrupted to accommodate the spatial requirements of pre-mRNA substrate recognition and catalysis. Splicing activators and repressors, such as SR proteins and hnRNPs, modulate spliceosome assembly and regulate alternative splicing. However, it remains unclear how they differentially interact with the core spliceosome to perform their functions.
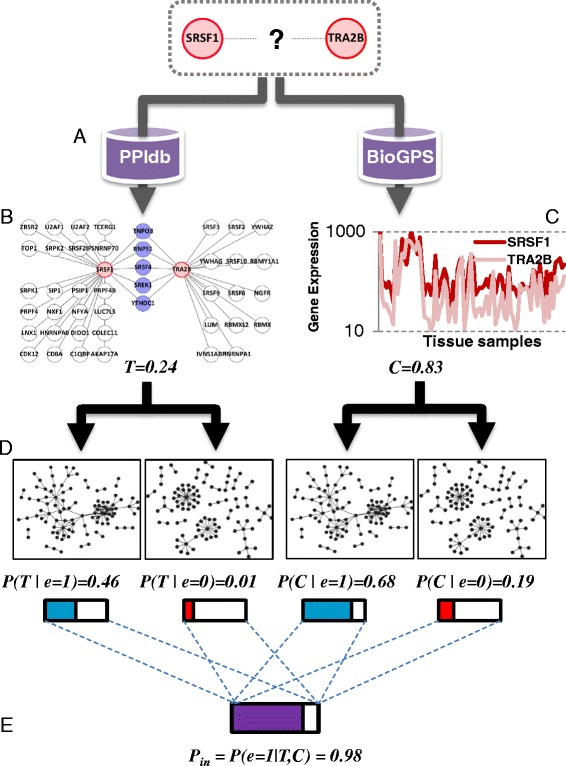
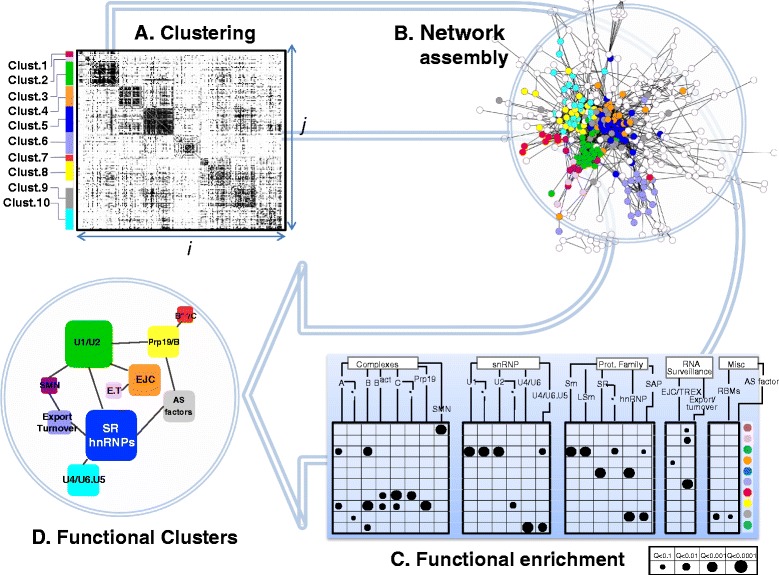
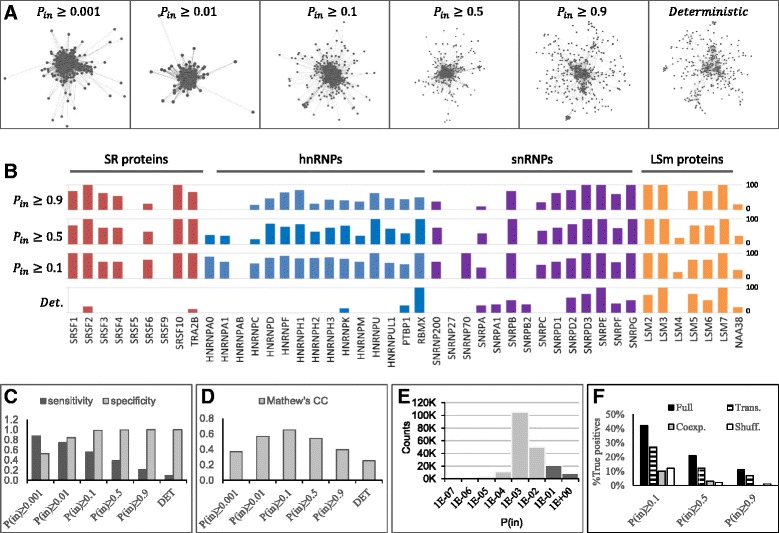
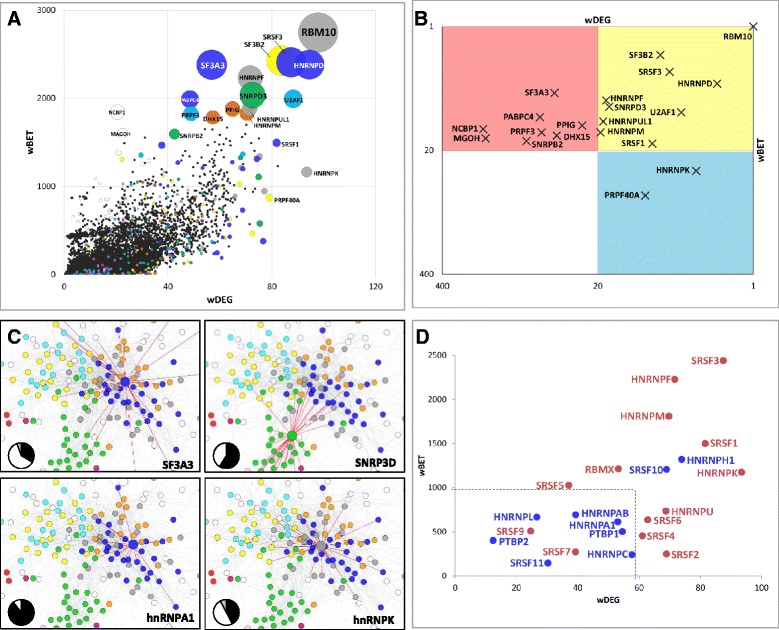
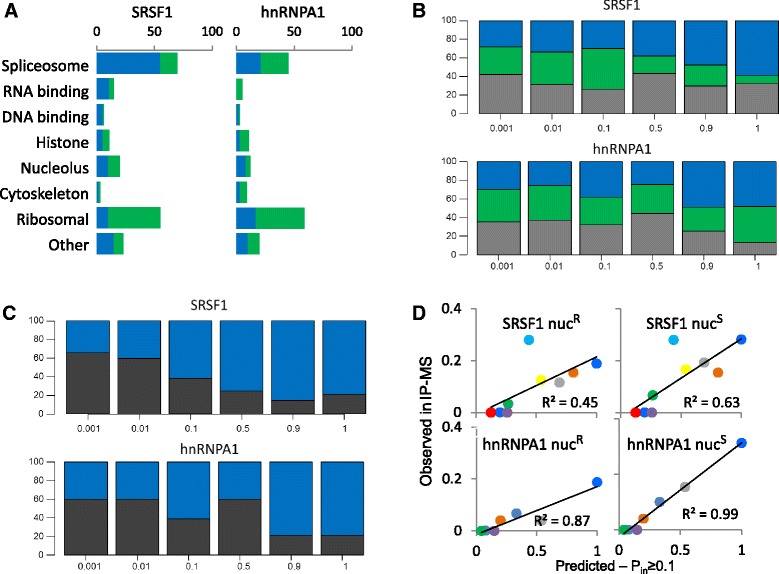
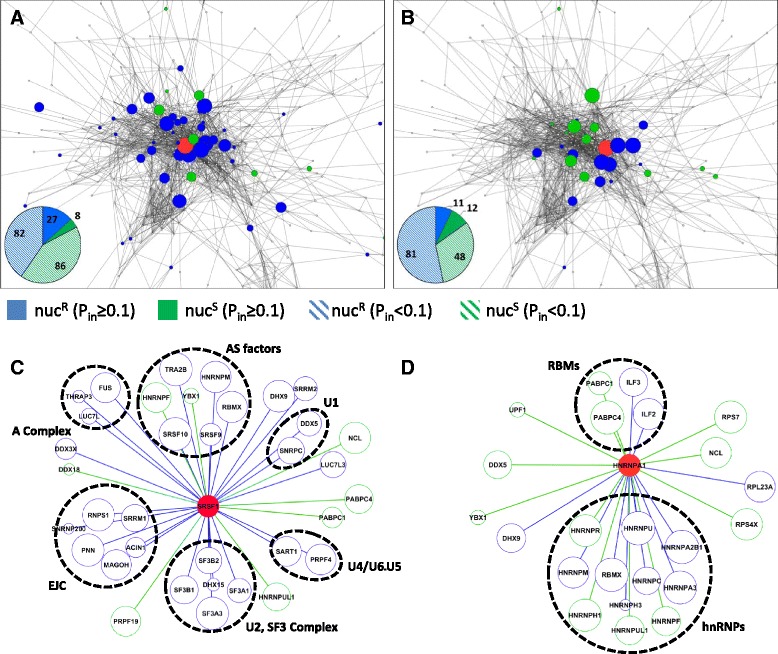
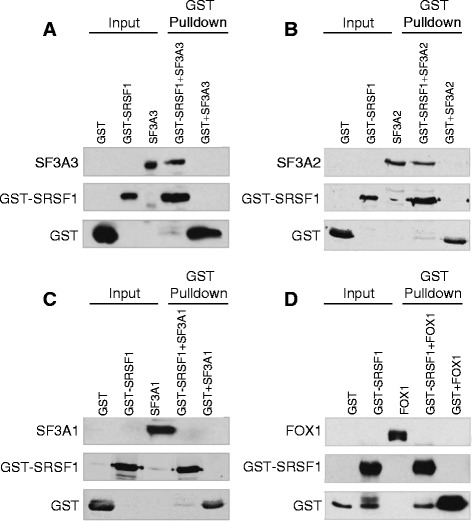
Results: Here, we investigate the protein connectivity of SR and hnRNP proteins to the core spliceosome using probabilistic network reconstruction based on the integration of interactome and gene expression data. We validate our model by immunoprecipitation and mass spectrometry of the prototypical splicing factors SRSF1 and hnRNPA1. Network analysis reveals that a factor's properties as an activator or repressor can be predicted from its overall connectivity to the rest of the spliceosome. In addition, we discover and experimentally validate PPIs between the oncoprotein SRSF1 and members of the anti-tumor drug target SF3 complex. Our findings suggest that activators promote the formation of PPIs between spliceosomal sub-complexes, whereas repressors mostly operate through protein-RNA interactions.
Conclusions: This study demonstrates that combining in-silico modeling with biochemistry can significantly advance the understanding of structure and function relationships in the human spliceosome.
Figures
Comment in
-
Interactome analysis brings splicing into focus.Genome Biol. 2015 Jul 7;16(1):135. doi: 10.1186/s13059-015-0707-0. Genome Biol. 2015. PMID: 26149273 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
