

Genotype-phenotype correlation in neuronal migration disorders and cortical dysplasias
- PMID: 26052266
- PMCID: PMC4439546
- DOI: 10.3389/fnins.2015.00181
Genotype-phenotype correlation in neuronal migration disorders and cortical dysplasias
Abstract
Neuronal migration disorders are human (or animal) diseases that result from a disruption in the normal movement of neurons from their original birth site to their final destination during early development. As a consequence, the neurons remain somewhere along their migratory route, their location depending on the pathological mechanism and its severity. The neurons form characteristic abnormalities, which are morphologically classified into several types, such as lissencephaly, heterotopia, and cobblestone dysplasia. Polymicrogyria is classified as a group of malformations that appear secondary to post-migration development; however, recent findings of the underlying molecular mechanisms reveal overlapping processes in the neuronal migration and post-migration development stages. Mutations of many genes are involved in neuronal migration disorders, such as LIS1 and DCX in classical lissencephaly spectrum, TUBA1A in microlissencephaly with agenesis of the corpus callosum, and RELN and VLDLR in lissencephaly with cerebellar hypoplasia. ARX is of particular interest from basic and clinical perspectives because it is critically involved in tangential migration of GABAergic interneurons in the forebrain and its mutations cause a variety of phenotypes ranging from hydranencephaly or lissencephaly to early-onset epileptic encephalopathies, including Ohtahara syndrome and infantile spasms or intellectual disability with no brain malformations. The recent advances in gene and genome analysis technologies will enable the genetic basis of neuronal migration disorders to be unraveled, which, in turn, will facilitate genotype-phenotype correlations to be determined.
Keywords: ARX; DCX; LIS1; heterotopia; interneuronopathy; lissencephaly; polymicrogyria; tubulinopathy.
Figures
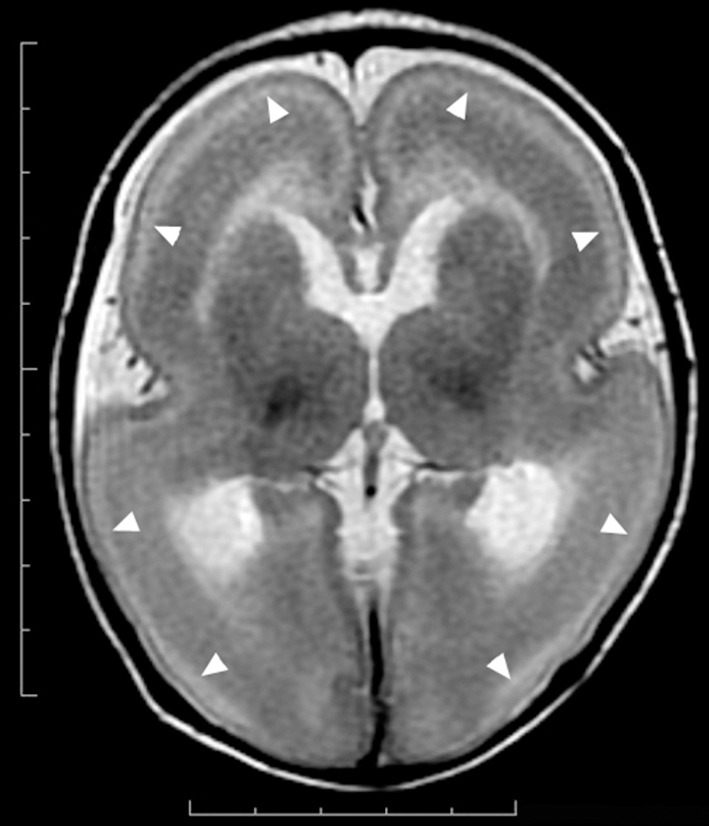
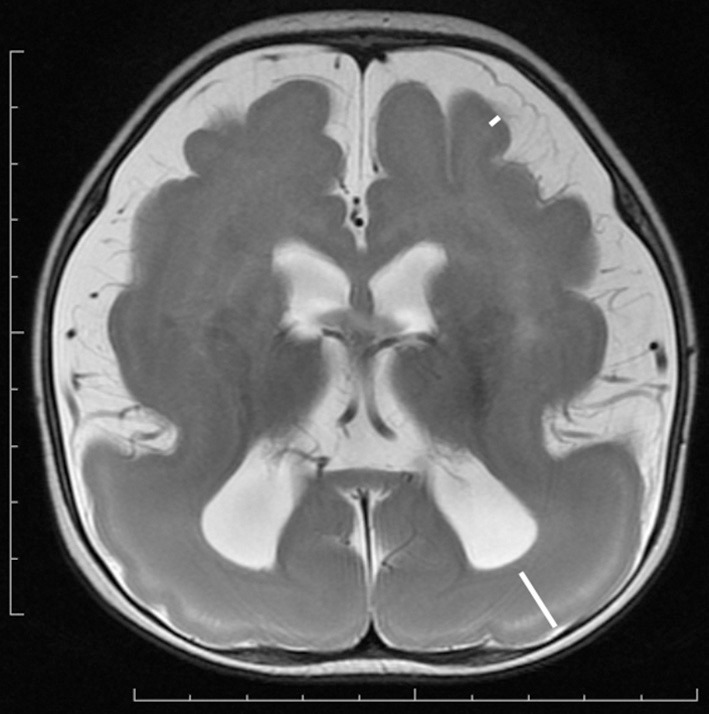
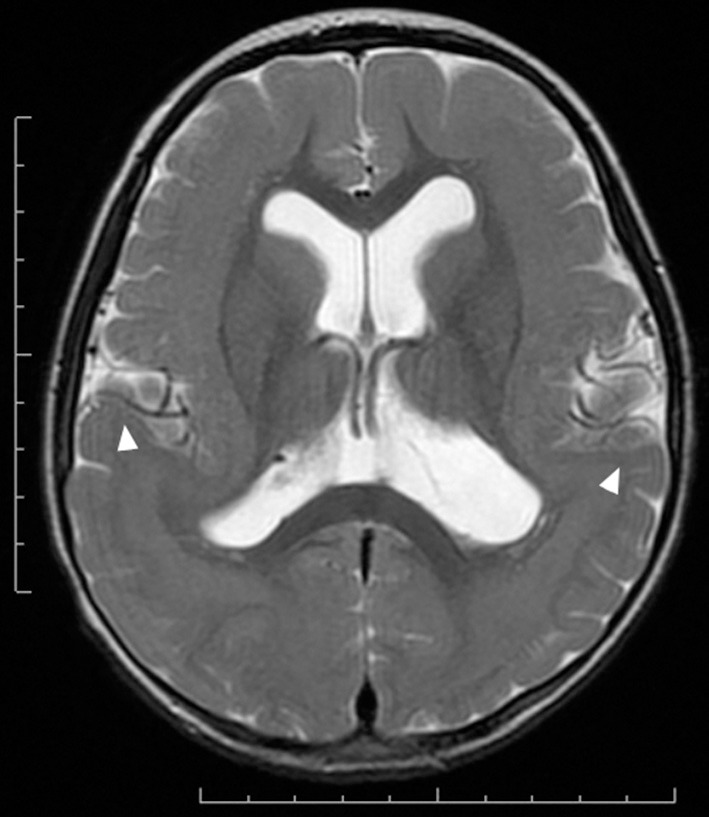


References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous