

Global circulation patterns of seasonal influenza viruses vary with antigenic drift
- PMID: 26053121
- PMCID: PMC4499780
- DOI: 10.1038/nature14460
Global circulation patterns of seasonal influenza viruses vary with antigenic drift
Abstract
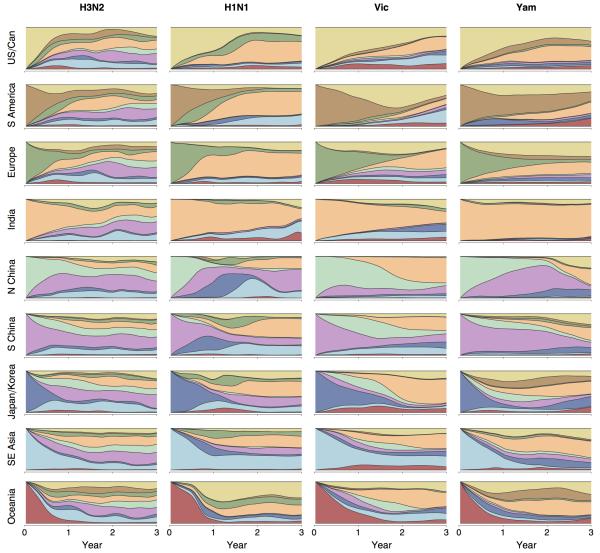
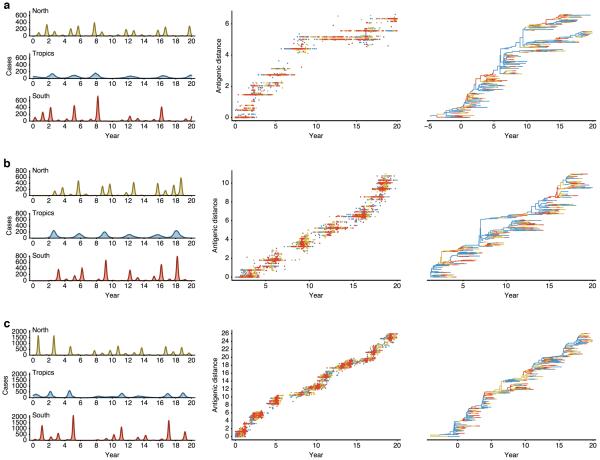
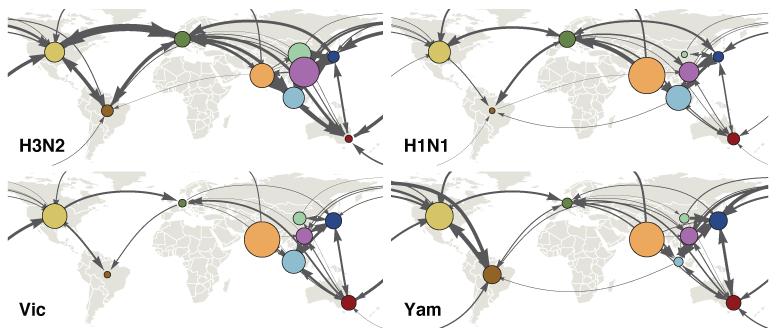
Understanding the spatiotemporal patterns of emergence and circulation of new human seasonal influenza virus variants is a key scientific and public health challenge. The global circulation patterns of influenza A/H3N2 viruses are well characterized, but the patterns of A/H1N1 and B viruses have remained largely unexplored. Here we show that the global circulation patterns of A/H1N1 (up to 2009), B/Victoria, and B/Yamagata viruses differ substantially from those of A/H3N2 viruses, on the basis of analyses of 9,604 haemagglutinin sequences of human seasonal influenza viruses from 2000 to 2012. Whereas genetic variants of A/H3N2 viruses did not persist locally between epidemics and were reseeded from East and Southeast Asia, genetic variants of A/H1N1 and B viruses persisted across several seasons and exhibited complex global dynamics with East and Southeast Asia playing a limited role in disseminating new variants. The less frequent global movement of influenza A/H1N1 and B viruses coincided with slower rates of antigenic evolution, lower ages of infection, and smaller, less frequent epidemics compared to A/H3N2 viruses. Detailed epidemic models support differences in age of infection, combined with the less frequent travel of children, as probable drivers of the differences in the patterns of global circulation, suggesting a complex interaction between virus evolution, epidemiology, and human behaviour.
Figures








References
-
- Russell CA, et al. The global circulation of seasonal influenza A (H3N2) viruses. Science. 2008;320:340–346. - PubMed
Publication types
MeSH terms
Grants and funding
- 093488/Z/10/Z/WT_/Wellcome Trust/United Kingdom
- MC_U117585868/MRC_/Medical Research Council/United Kingdom
- 093488/WT_/Wellcome Trust/United Kingdom
- R01 TW008246/TW/FIC NIH HHS/United States
- U01 GM110721-01/GM/NIGMS NIH HHS/United States
- U117512723/MRC_/Medical Research Council/United Kingdom
- MC_U117512723/MRC_/Medical Research Council/United Kingdom
- 260864/ERC_/European Research Council/International
- U54 GM111274/GM/NIGMS NIH HHS/United States
- U01 GM110721/GM/NIGMS NIH HHS/United States
- R01 AI107034/AI/NIAID NIH HHS/United States
- MR/J008761/1/MRC_/Medical Research Council/United Kingdom
- R01 AI 107034/AI/NIAID NIH HHS/United States
- R01 TW008246-01/TW/FIC NIH HHS/United States
- MR/K010174/1/MRC_/Medical Research Council/United Kingdom
- U51 IP000333/IP/NCIRD CDC HHS/United States
- 095831/WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
