

Recurrent somatic mutations in regulatory regions of human cancer genomes
- PMID: 26053494
- PMCID: PMC4485503
- DOI: 10.1038/ng.3332
Recurrent somatic mutations in regulatory regions of human cancer genomes
Abstract
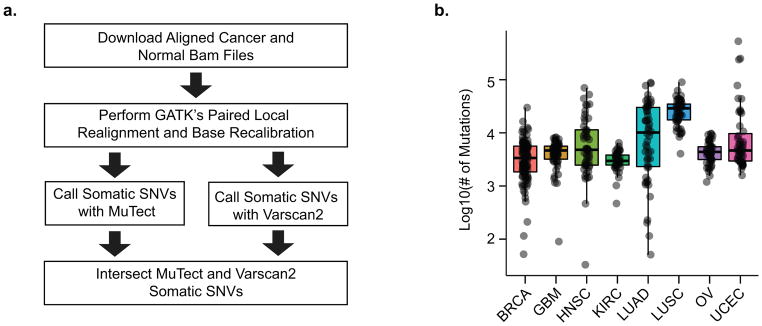
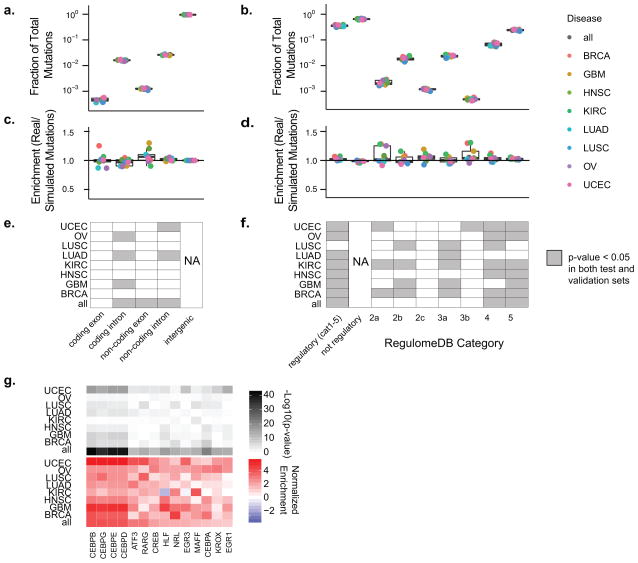
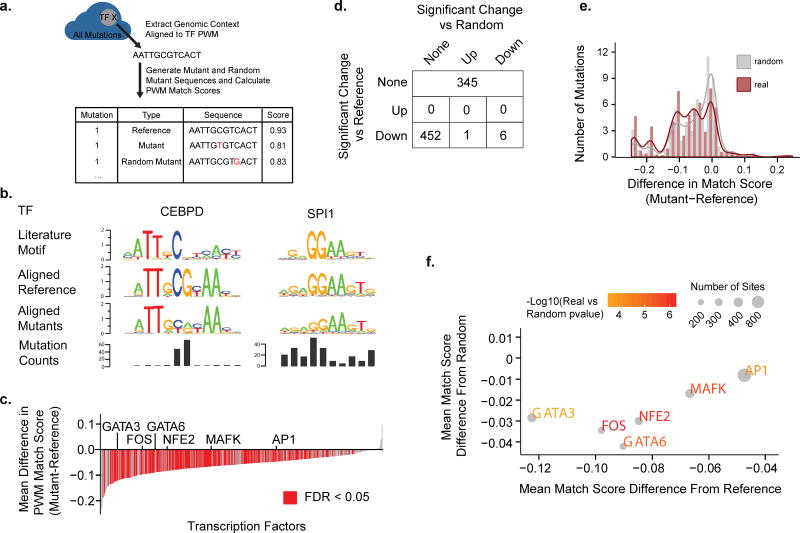
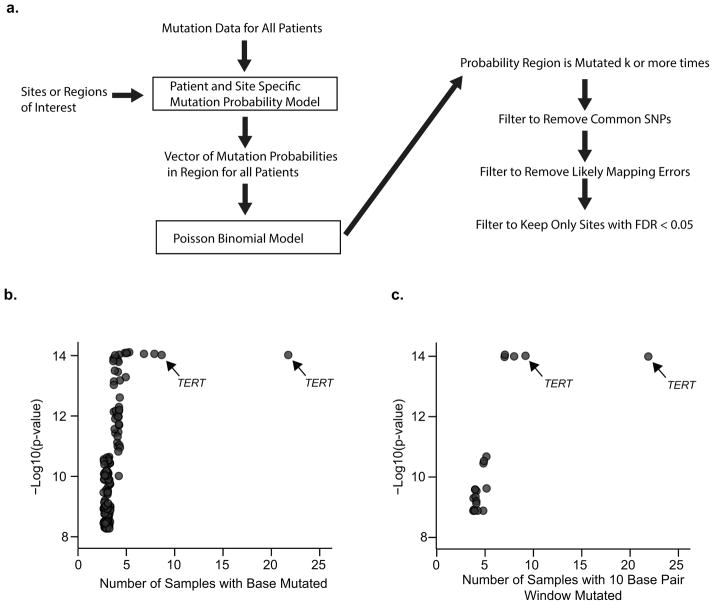
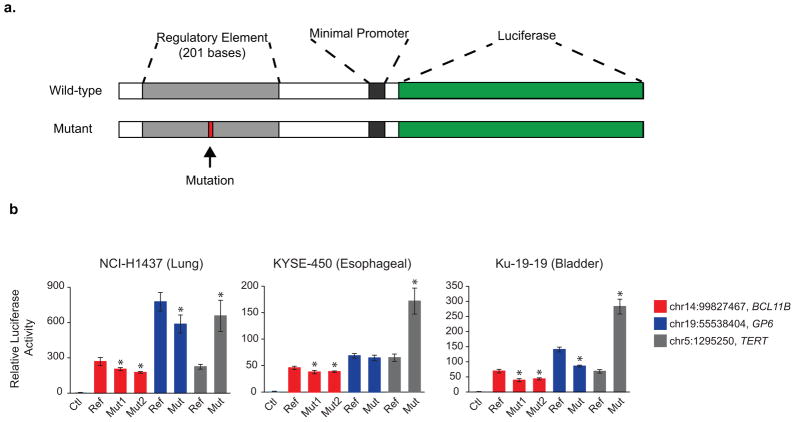
Aberrant regulation of gene expression in cancer can promote survival and proliferation of cancer cells. Here we integrate whole-genome sequencing data from The Cancer Genome Atlas (TCGA) for 436 patients from 8 cancer subtypes with ENCODE and other regulatory annotations to identify point mutations in regulatory regions. We find evidence for positive selection of mutations in transcription factor binding sites, consistent with these sites regulating important cancer cell functions. Using a new method that adjusts for sample- and genomic locus-specific mutation rates, we identify recurrently mutated sites across individuals with cancer. Mutated regulatory sites include known sites in the TERT promoter and many new sites, including a subset in proximity to cancer-related genes. In reporter assays, two new sites display decreased enhancer activity upon mutation. These data demonstrate that many regulatory regions contain mutations under selective pressure and suggest a greater role for regulatory mutations in cancer than previously appreciated.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Hoyert DL, Xu J. Deaths: Preliminary Data for 2011. National Vital Statistics Reports. 2012;61 - PubMed
-
- Howlander, et al. SEER Cancer Statistics Review, 1975–2010. 2013 at < http://seer.cancer.gov/csr/1975_2010/>.
-
-
Lifetime Risk (Percent) of Being Diagnosed with Cancer by Site and Race/Ethnicity: Males, 18 SEER Areas, 2008–2010 (Table 1.16) and Females, 18 SEER Areas, 2008–2010 (Table 1.17). at <
http://seer.cancer.gov/csr/1975_2010/results_merged/topic_lifetime_risk_diagnosis.pdf >
-
-
-
Lifetime Risk (Percent) of Dying from Cancer by Site and Race/Ethnicity: Males, Total US, 2008–2010 (Table 1.19) and Females, Total US, 2008–2010 (Table 1.20). at <
http://seer.cancer.gov/csr/1975_2010/results_merged/topic_lifetime_risk_death.pdf >
-
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
