

Whole-genome fingerprint of the DNA methylome during human B cell differentiation
- PMID: 26053498
- PMCID: PMC5444519
- DOI: 10.1038/ng.3291
Whole-genome fingerprint of the DNA methylome during human B cell differentiation
Abstract
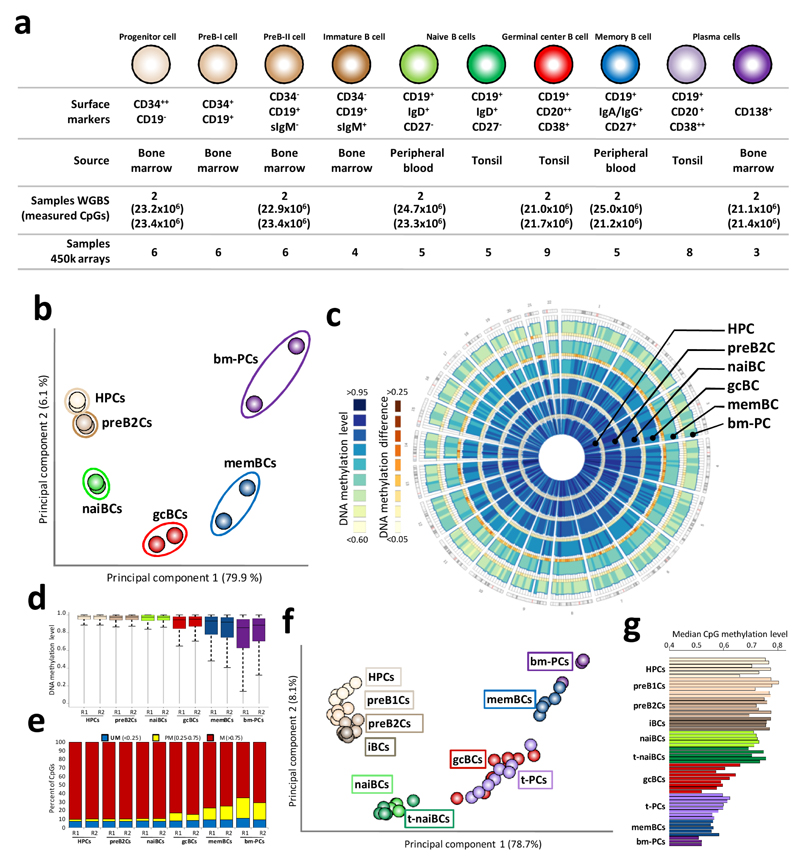
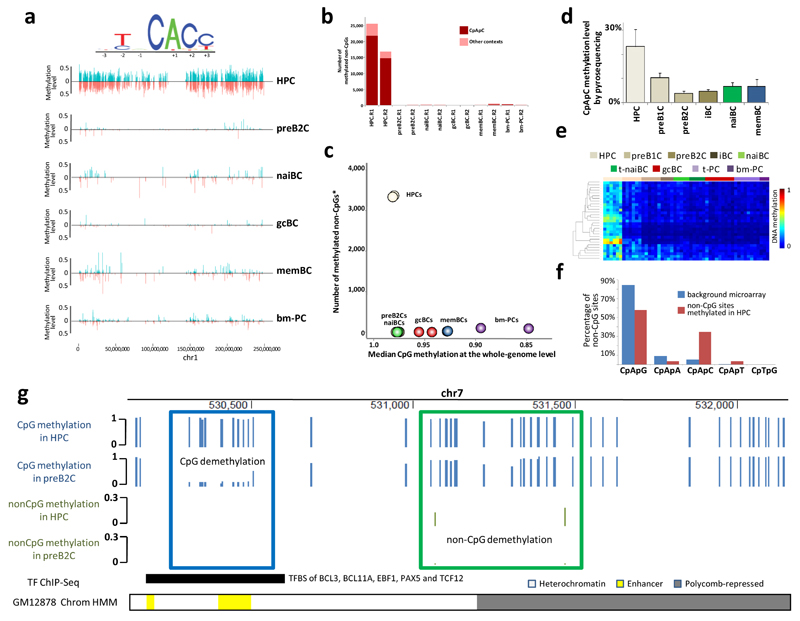
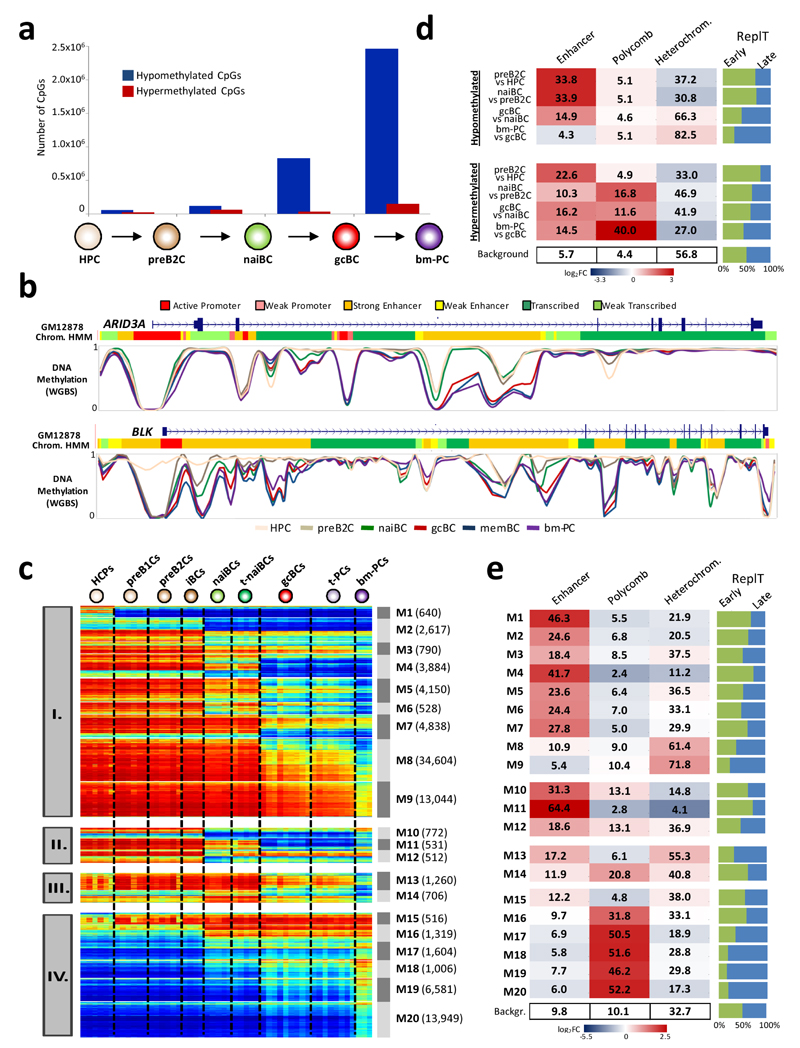
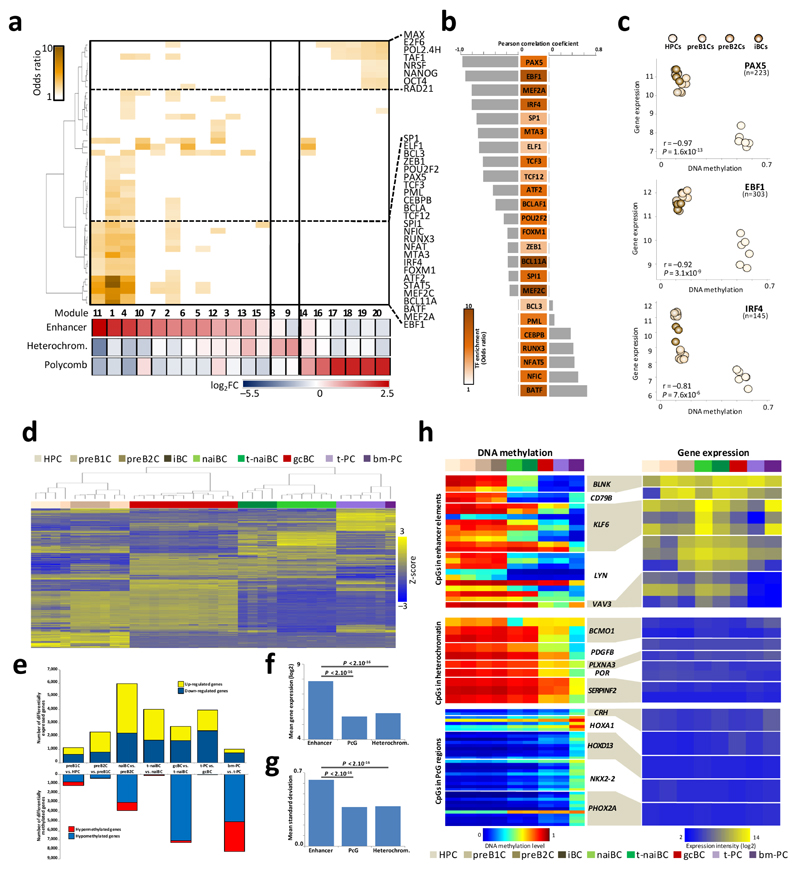
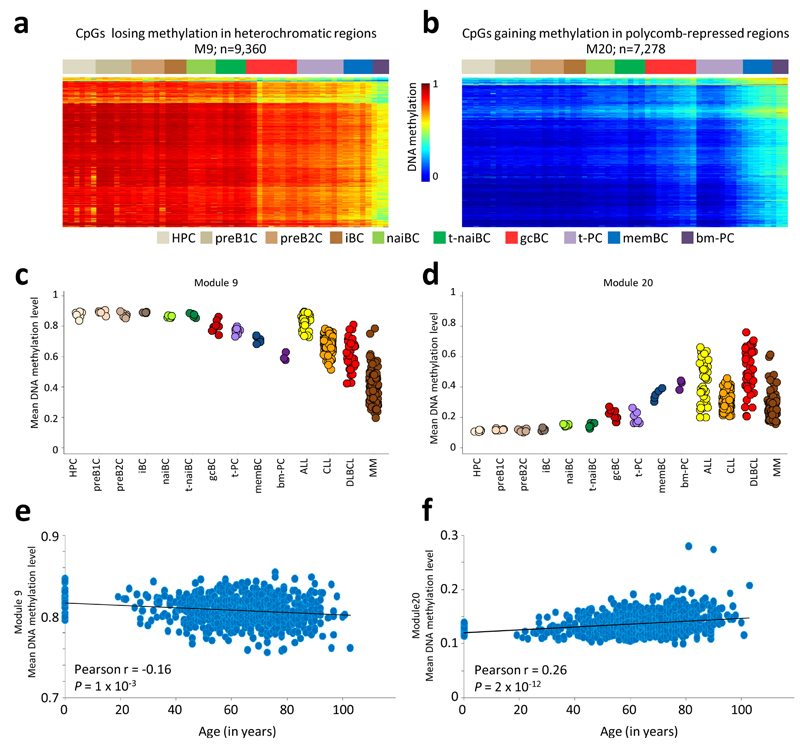
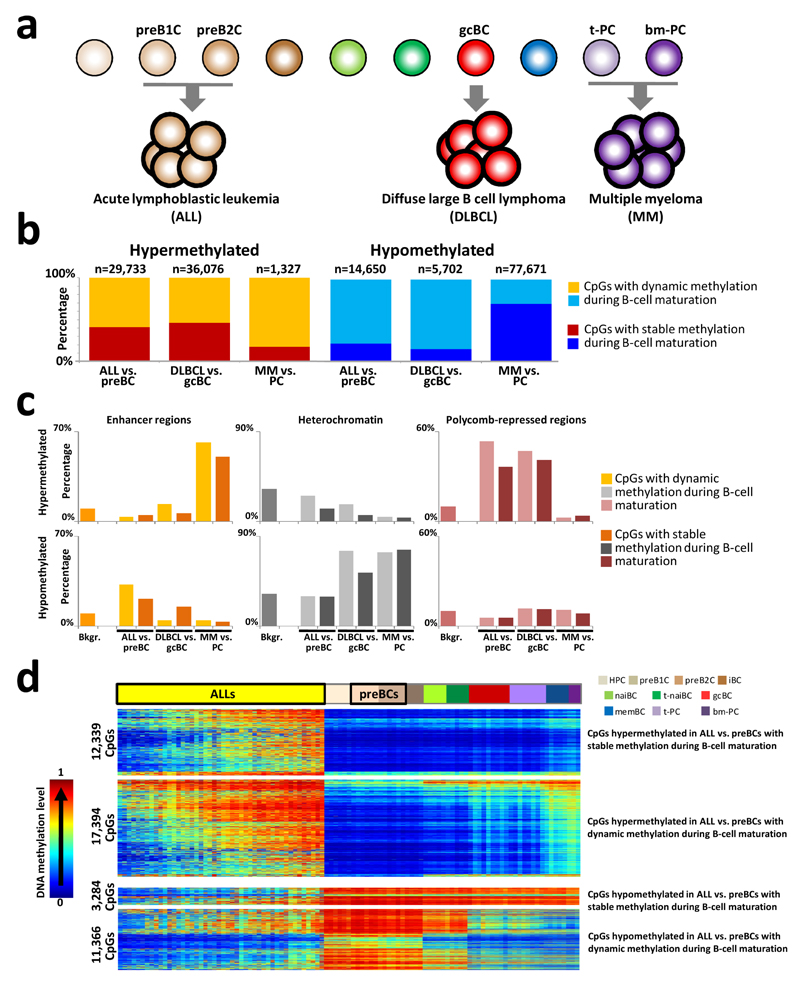
We analyzed the DNA methylome of ten subpopulations spanning the entire B cell differentiation program by whole-genome bisulfite sequencing and high-density microarrays. We observed that non-CpG methylation disappeared upon B cell commitment, whereas CpG methylation changed extensively during B cell maturation, showing an accumulative pattern and affecting around 30% of all measured CpG sites. Early differentiation stages mainly displayed enhancer demethylation, which was associated with upregulation of key B cell transcription factors and affected multiple genes involved in B cell biology. Late differentiation stages, in contrast, showed extensive demethylation of heterochromatin and methylation gain at Polycomb-repressed areas, and genes with apparent functional impact in B cells were not affected. This signature, which has previously been linked to aging and cancer, was particularly widespread in mature cells with an extended lifespan. Comparing B cell neoplasms with their normal counterparts, we determined that they frequently acquire methylation changes in regions already undergoing dynamic methylation during normal B cell differentiation.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell. 2007;128:669–81. - PubMed
-
- Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013;14:204–20. - PubMed
-
- Bergman Y, Cedar H. DNA methylation dynamics in health and disease. Nat Struct Mol Biol. 2013;20:274–81. - PubMed
-
- Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
