

Autophagy in autoimmune disease
- PMID: 26054920
- PMCID: PMC4486076
- DOI: 10.1007/s00109-015-1297-8
Autophagy in autoimmune disease
Abstract
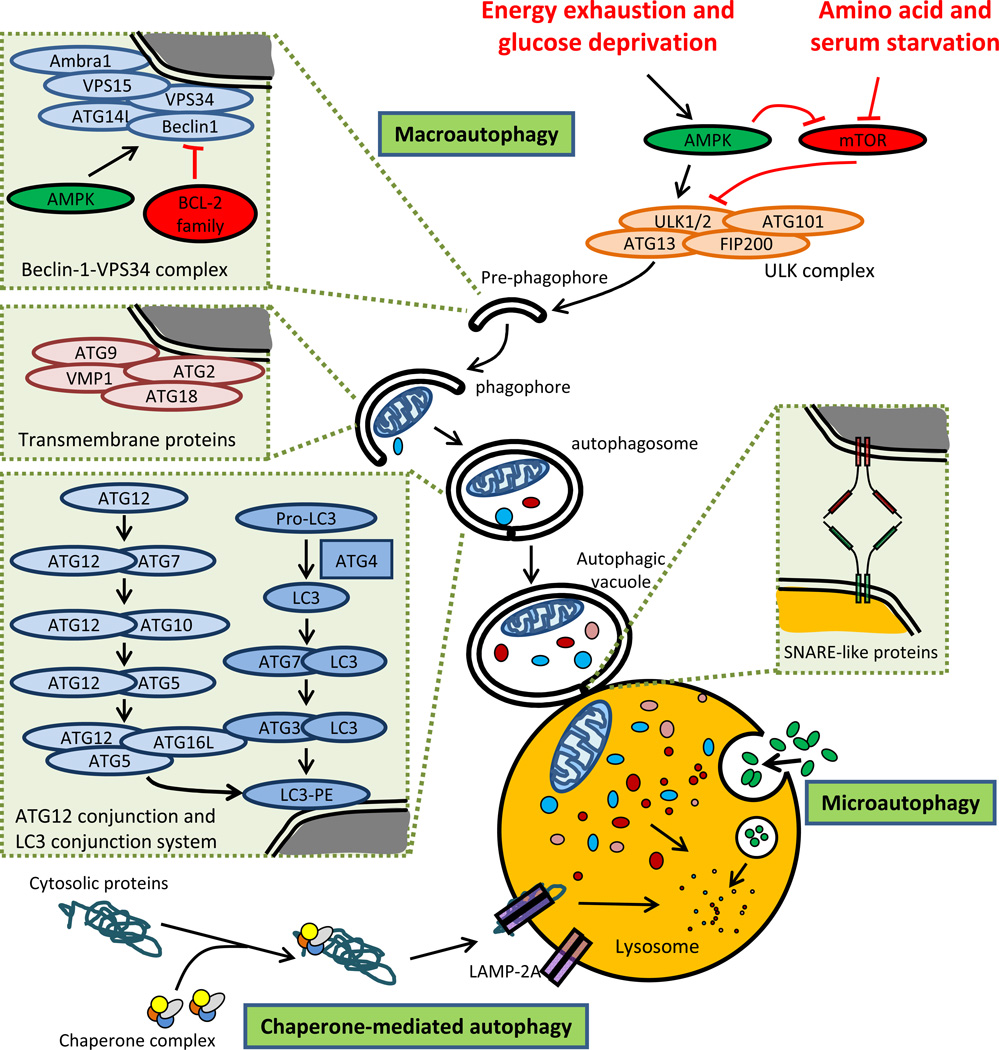
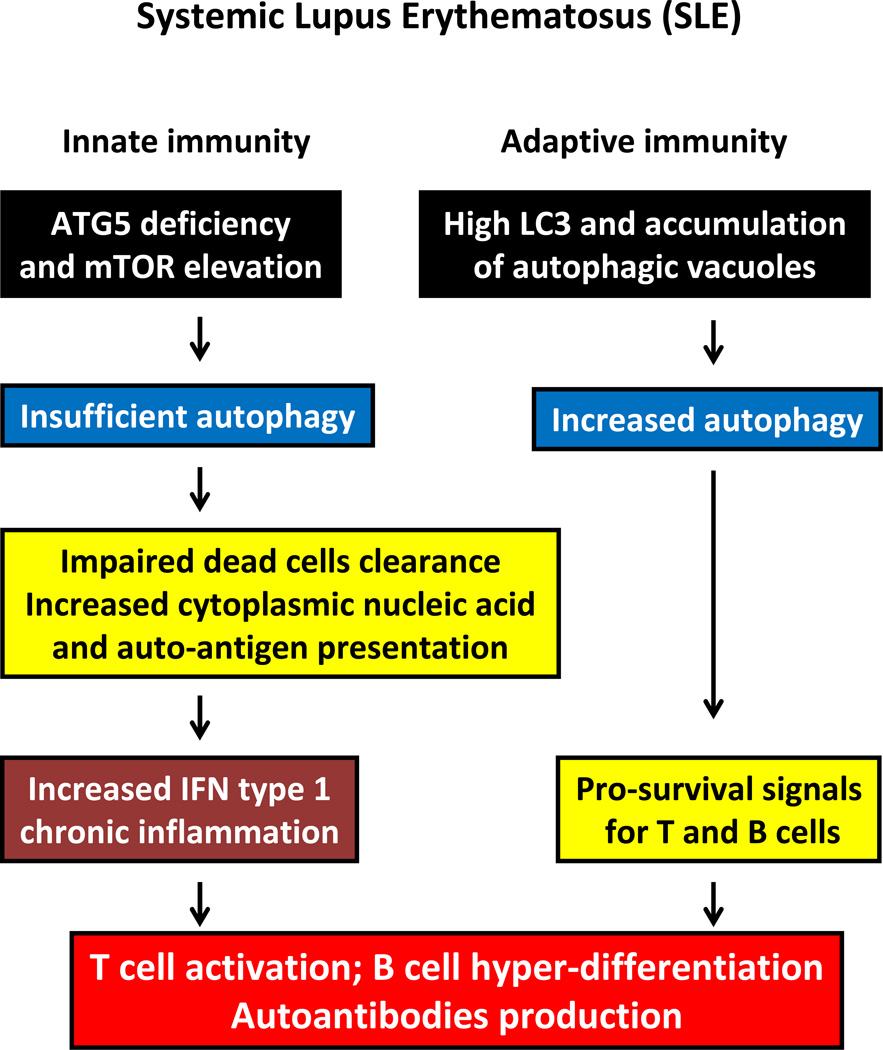
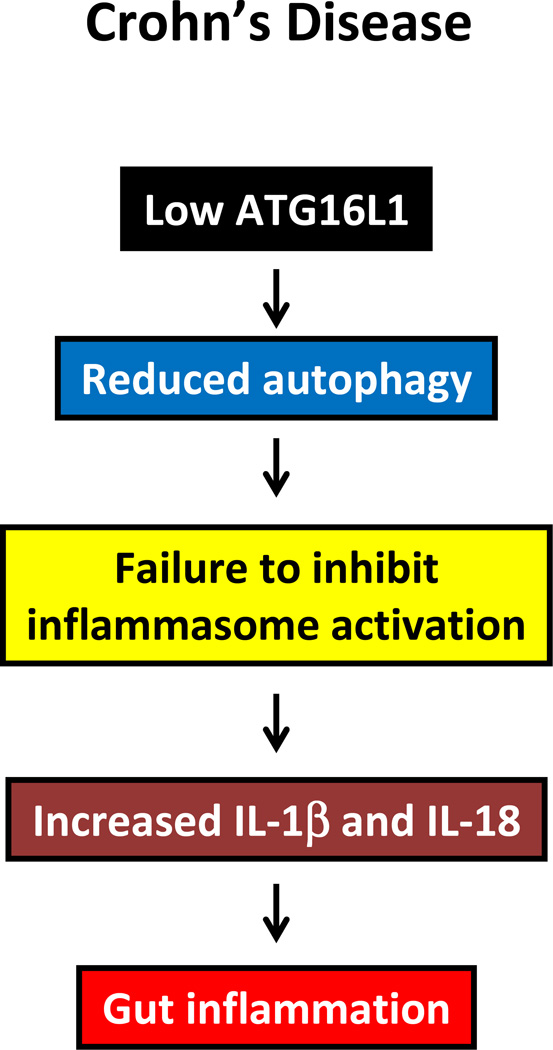
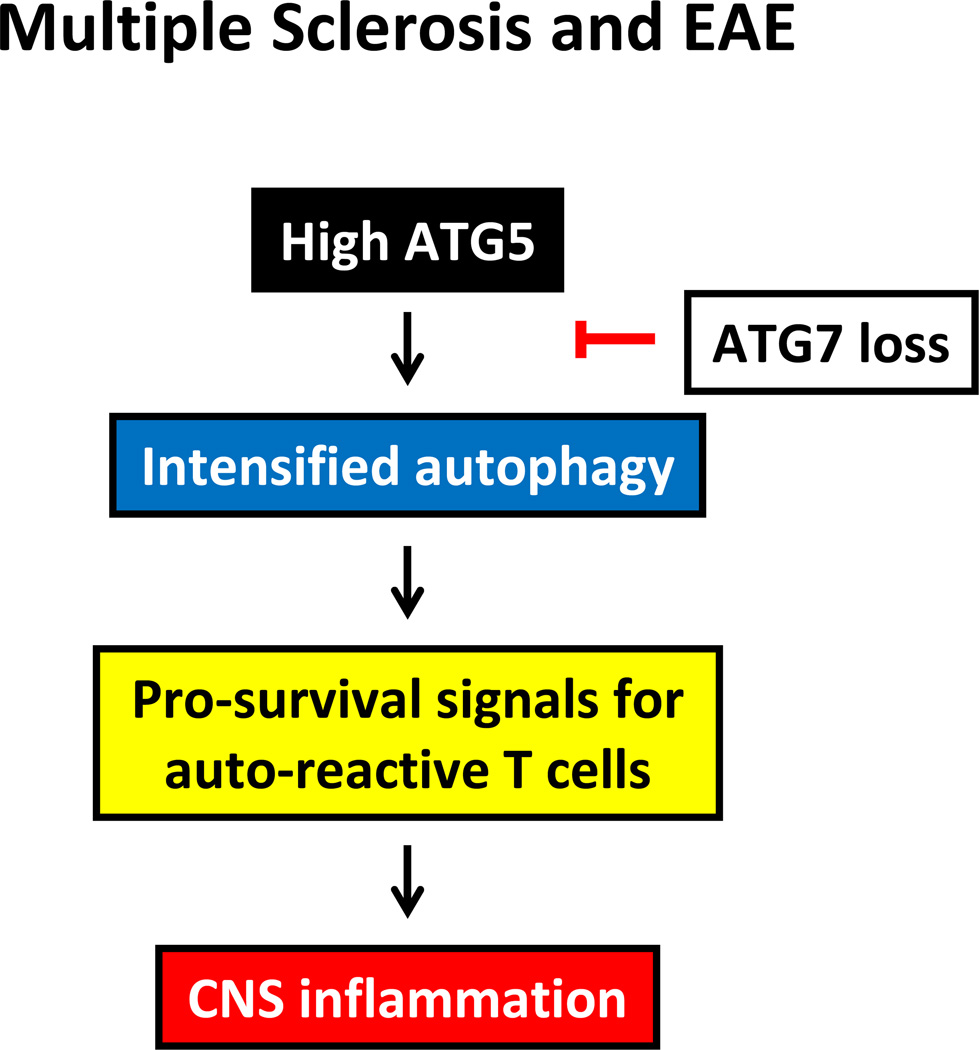
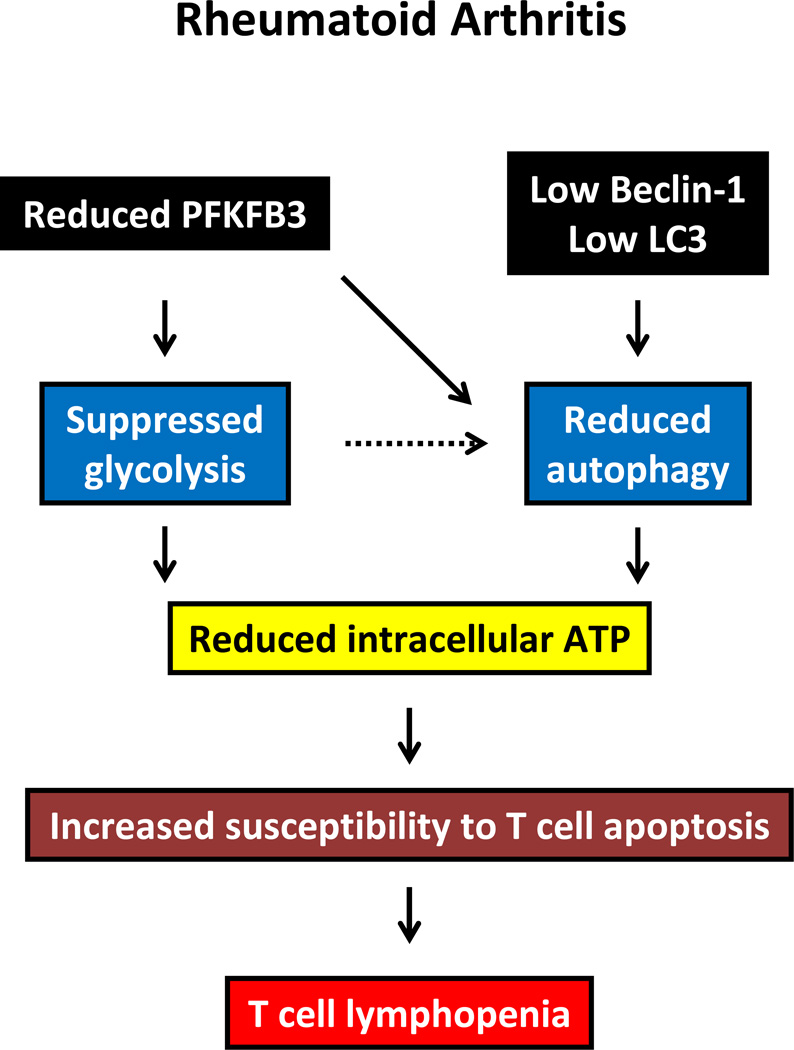
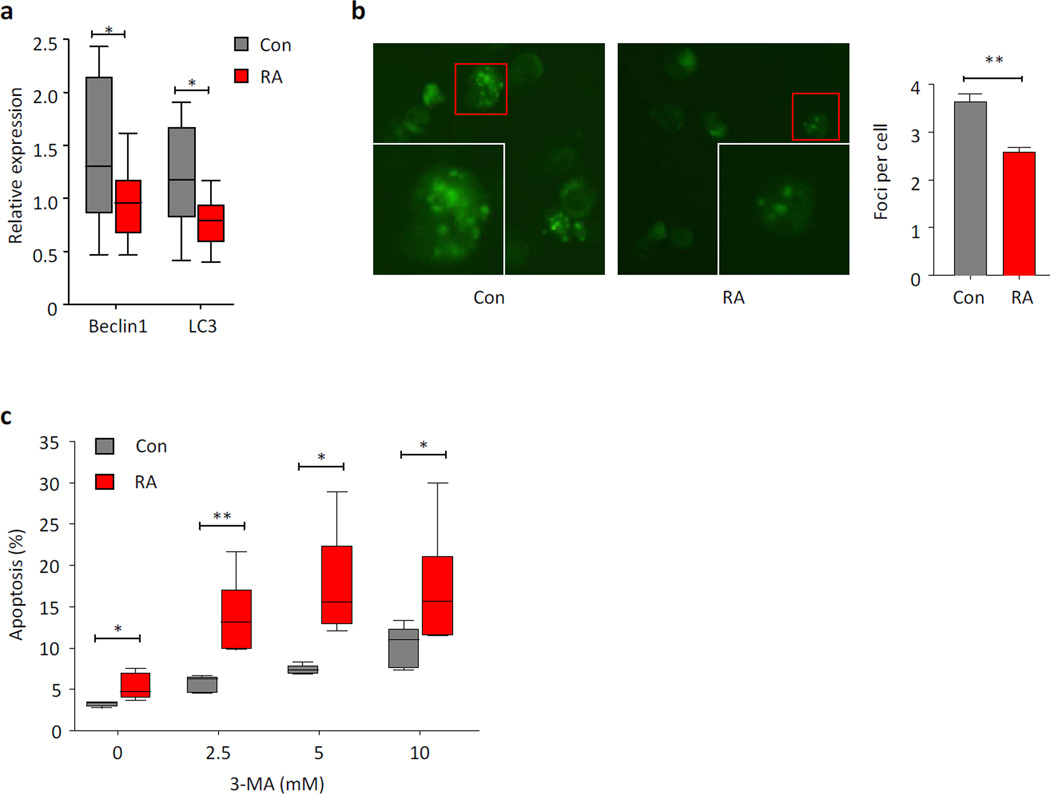
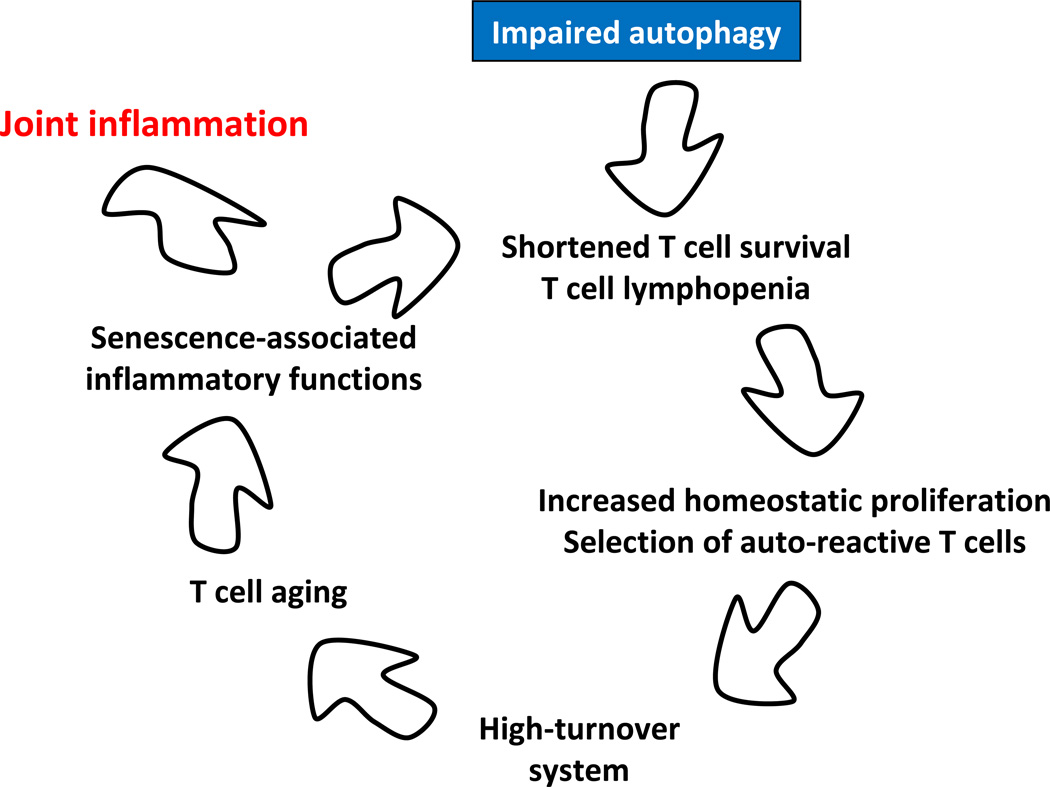
Autophagy is a protective and life-sustaining process in which cytoplasmic components are packaged into double-membrane vesicles and targeted to lysosomes for degradation. This process of cellular self-digestion is an essential stress response and is cytoprotective by removing damaged organelles and proteins that threaten the cell's survival. Key outcomes include energy generation and recycling of metabolic precursors. In the immune system, autophagy regulates processes such as antigen uptake and presentation, removal of pathogens, survival of short- and long-lived immune cells, and cytokine-dependent inflammation. In all cases, a window of optimal autophagic activity appears critical to balance catabolic, reparative, and inflammation-inducing processes. Dysregulation of autophagosome formation and autophagic flux can have deleterious consequences, ranging from a failure to "clean house" to the induction of autophagy-induced cell death. Abnormalities in the autophagic pathway have been implicated in numerous autoimmune diseases. Genome-wide association studies have linked polymorphisms in autophagy-related genes with predisposition for tissue-destructive inflammatory disease, specifically in inflammatory bowel disease and systemic lupus erythematosus. Although the precise mechanisms by which dysfunctional autophagy renders the host susceptible to continuous inflammation remain unclear, autophagy's role in regulating the long-term survival of adaptive immune cells has recently surfaced as a defect in multiple sclerosis and rheumatoid arthritis. Efforts are underway to identify autophagy-inducing and autophagy-suppressing pharmacologic interventions that can be added to immunosuppressive therapy to improve outcomes of patients with autoimmune disease.
Conflict of interest statement
Figures
References
-
- Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010;90:1383–1435. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
