

Structural basis for mutation-induced destabilization of profilin 1 in ALS
- PMID: 26056300
- PMCID: PMC4491777
- DOI: 10.1073/pnas.1424108112
Structural basis for mutation-induced destabilization of profilin 1 in ALS
Abstract
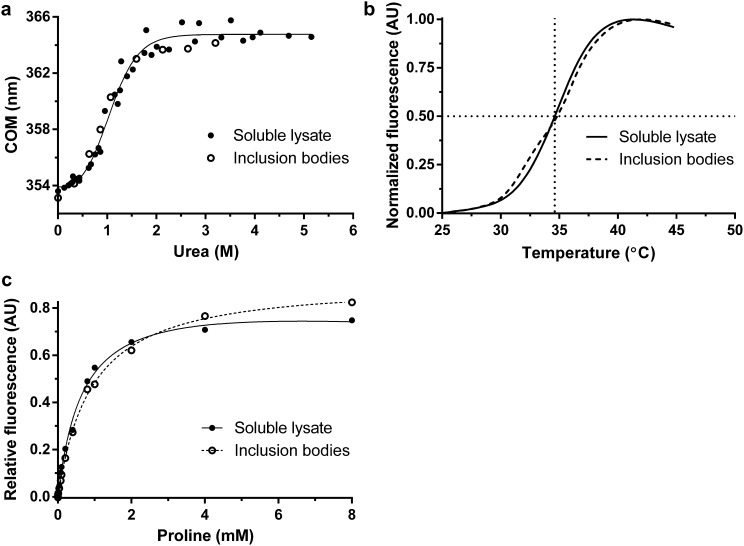
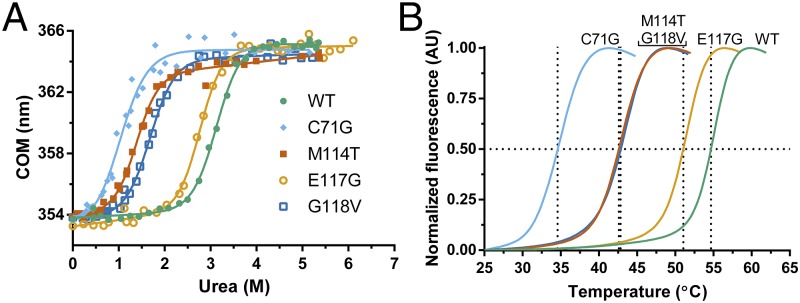
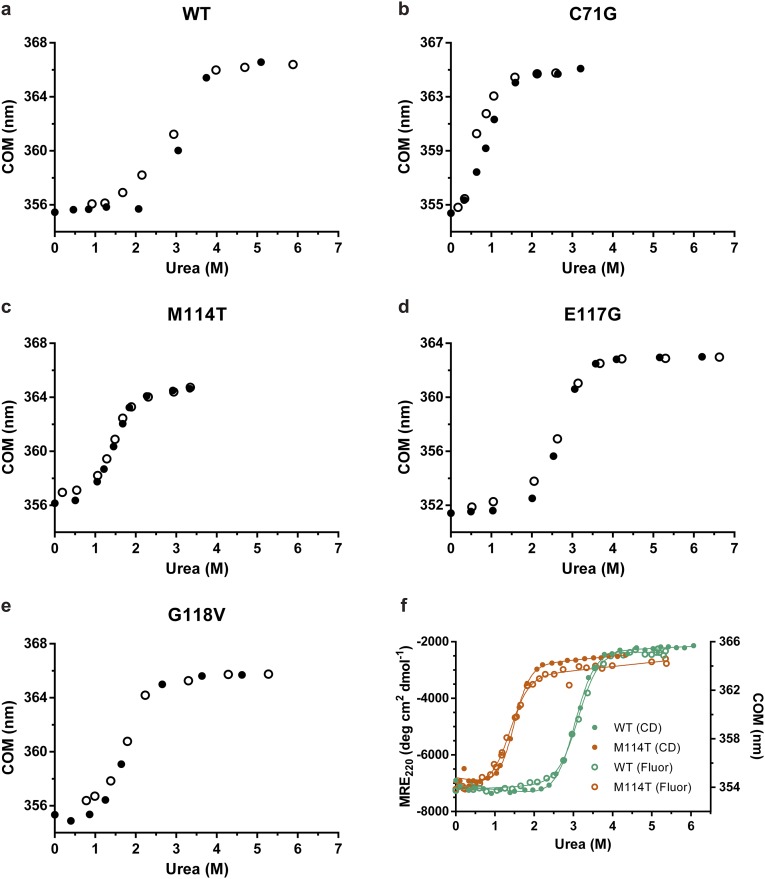
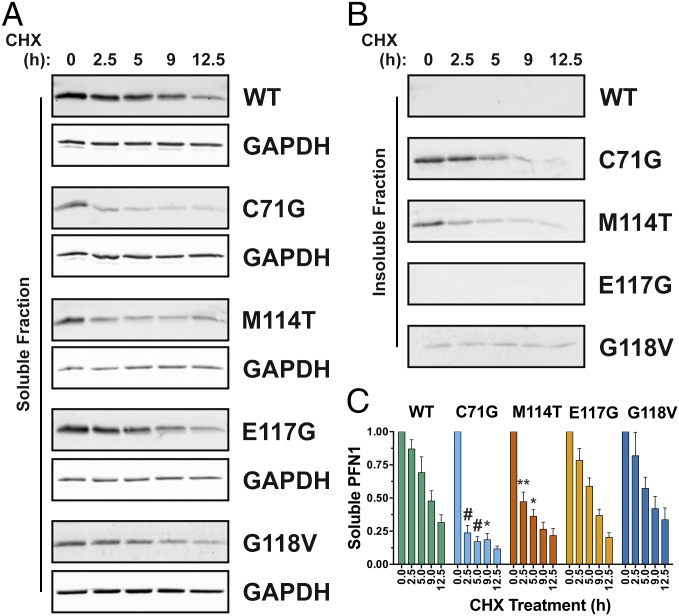
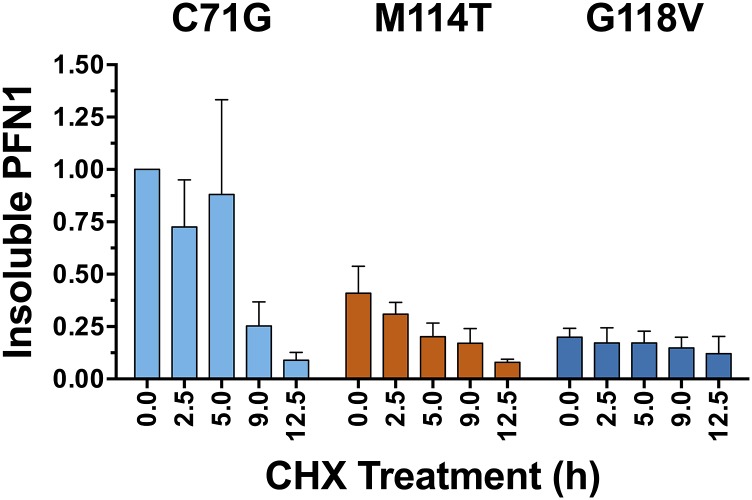
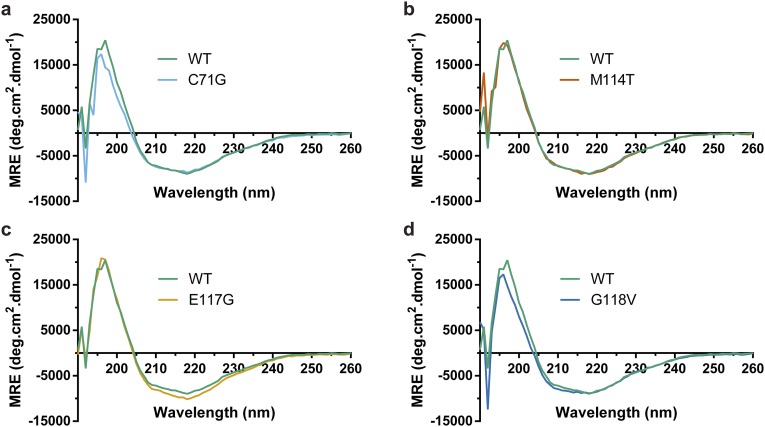
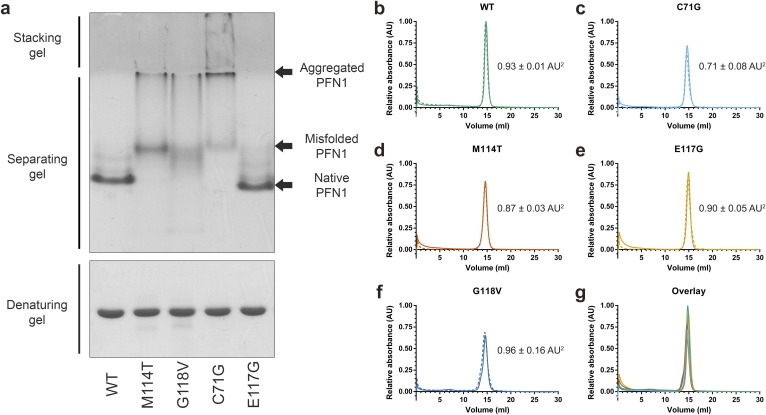
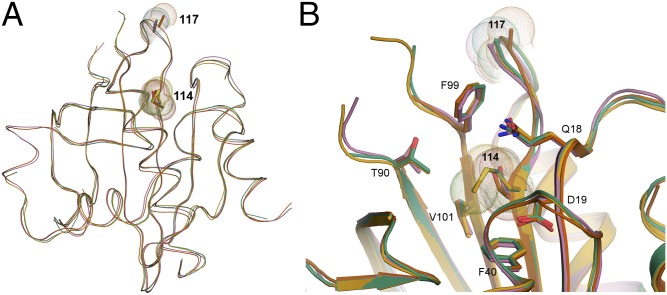
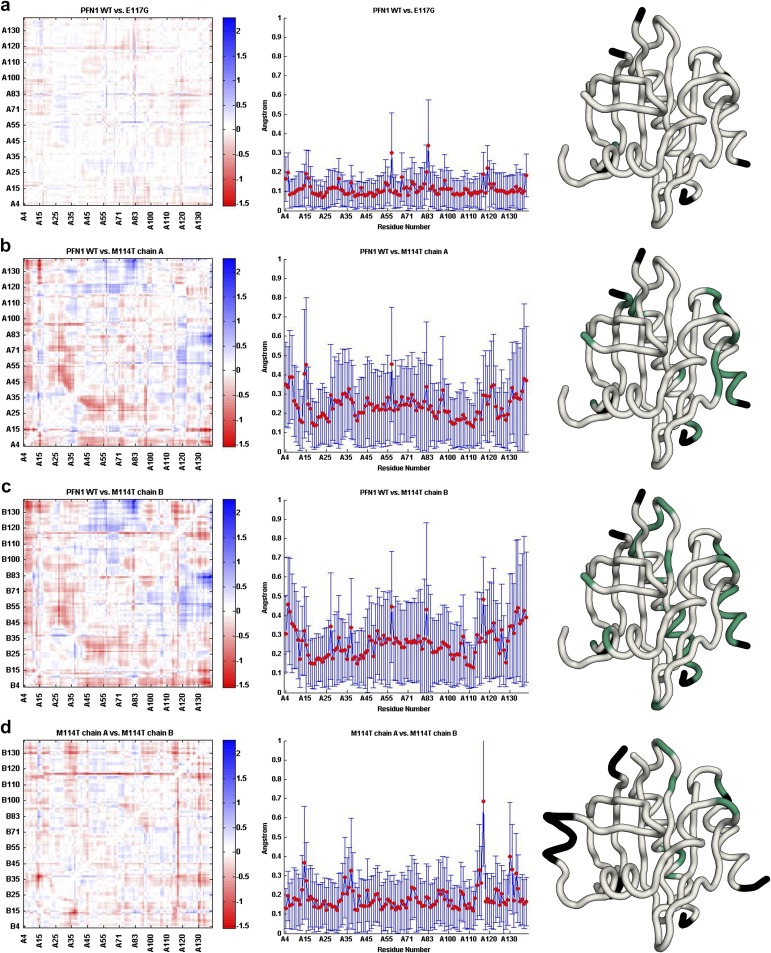
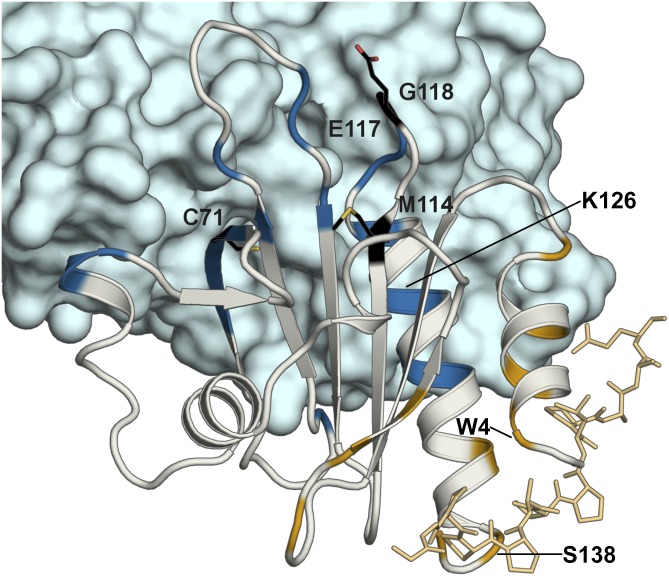
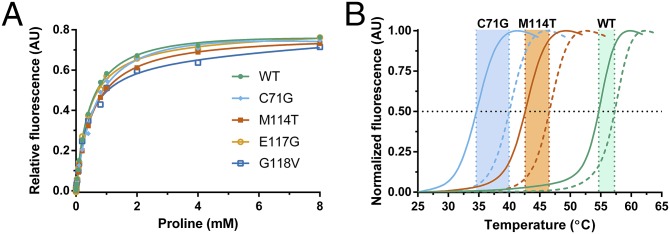
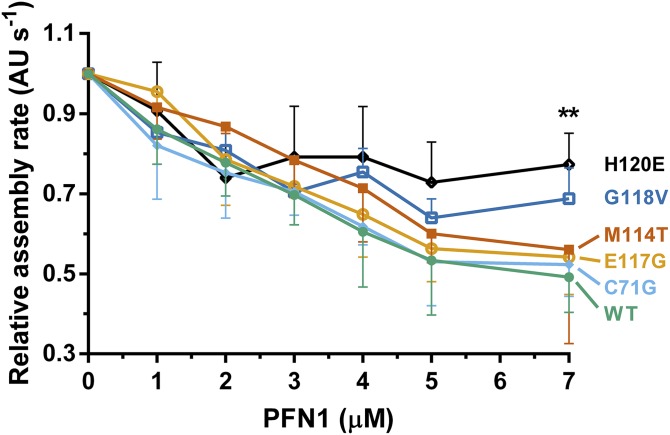
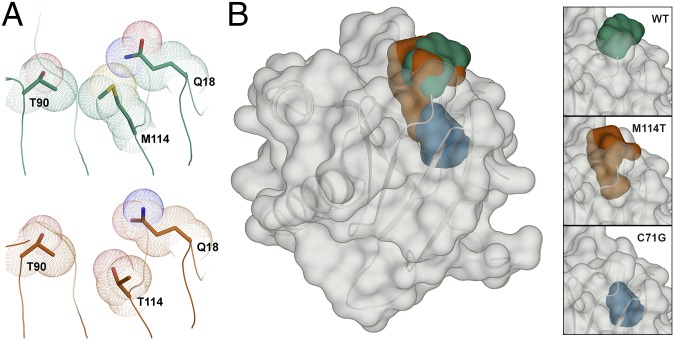
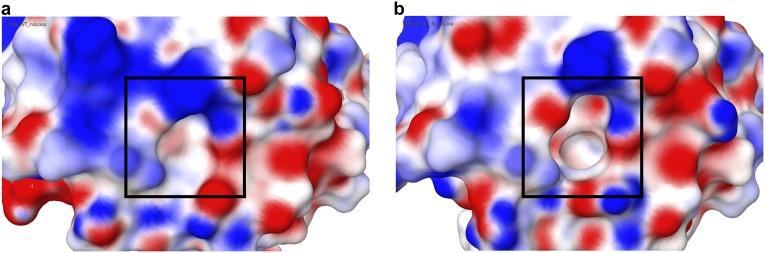
Mutations in profilin 1 (PFN1) are associated with amyotrophic lateral sclerosis (ALS); however, the pathological mechanism of PFN1 in this fatal disease is unknown. We demonstrate that ALS-linked mutations severely destabilize the native conformation of PFN1 in vitro and cause accelerated turnover of the PFN1 protein in cells. This mutation-induced destabilization can account for the high propensity of ALS-linked variants to aggregate and also provides rationale for their reported loss-of-function phenotypes in cell-based assays. The source of this destabilization is illuminated by the X-ray crystal structures of several PFN1 proteins, revealing an expanded cavity near the protein core of the destabilized M114T variant. In contrast, the E117G mutation only modestly perturbs the structure and stability of PFN1, an observation that reconciles the occurrence of this mutation in the control population. These findings suggest that a destabilized form of PFN1 underlies PFN1-mediated ALS pathogenesis.
Keywords: X-ray crystallography; amyotrophic lateral sclerosis; profilin 1; protein misfolding; protein stability.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Smith BN, et al. 2015. Novel mutations support a role for Profilin 1 in the pathogenesis of ALS. Neurobiol Aging 36(3):1602.e17–1602.e27.
-
- Witke W. The role of profilin complexes in cell motility and other cellular processes. Trends Cell Biol. 2004;14(8):461–469. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
