

Selective vulnerability of motoneuron and perturbed mitochondrial calcium homeostasis in amyotrophic lateral sclerosis: implications for motoneurons specific calcium dysregulation
- PMID: 26056593
- PMCID: PMC4452055
- DOI: 10.1186/2052-8426-2-26
Selective vulnerability of motoneuron and perturbed mitochondrial calcium homeostasis in amyotrophic lateral sclerosis: implications for motoneurons specific calcium dysregulation
Abstract
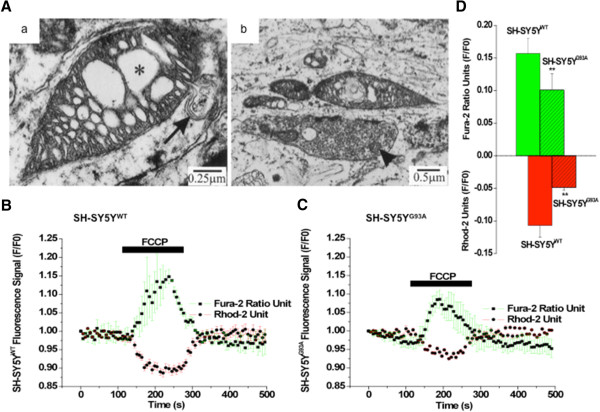
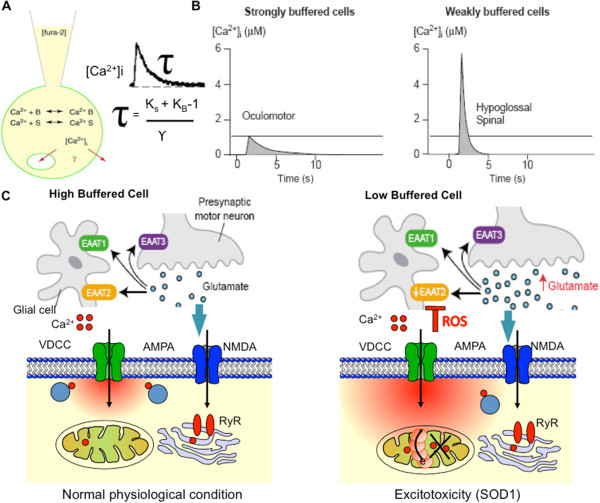
Amyotrophic lateral sclerosis (ALS) is a lethal neurodegenerative disorder characterized by the selective degeneration of defined subgroups of motoneuron in the brainstem, spinal cord and motor cortex with signature hallmarks of mitochondrial Ca(2+) overload, free radical damage, excitotoxicity and impaired axonal transport. Although intracellular disruptions of cytosolic and mitochondrial calcium, and in particular low cytosolic calcium ([Ca(2+)]c) buffering and a strong interaction between metabolic mechanisms and [Ca(2+)]i have been identified predominantly in motoneuron impairment, the causes of these disruptions are unknown. The existing evidence suggests that the mutant superoxide dismutase1 (mtSOD1)-mediated toxicity in ALS acts through mitochondria, and that alteration in cytosolic and mitochondria-ER microdomain calcium accumulation are critical to the neurodegenerative process. Furthermore, chronic excitotoxcity mediated by Ca(2+)-permeable AMPA and NMDA receptors seems to initiate vicious cycle of intracellular calcium dysregulation which leads to toxic Ca(2+) overload and thereby selective neurodegeneration. Recent advancement in the experimental analysis of calcium signals with high spatiotemporal precision has allowed investigations of calcium regulation in-vivo and in-vitro in different cell types, in particular selectively vulnerable/resistant cell types in different animal models of this motoneuron disease. This review provides an overview of latest advances in this field, and focuses on details of what has been learned about disrupted Ca(2+) homeostasis and mitochondrial degeneration. It further emphasizes the critical role of mitochondria in preventing apoptosis by acting as a Ca(2+) buffers, especially in motoneurons, in pathophysiological conditions such as ALS.
Keywords: Amyotrophic lateral sclerosis (ALS); Calcium buffering; Calcium dysregulation; ER-mitochondria calcium cycle (ERMCC); Mitochondria; Motoneuron; Multidrug therapy; Multifactorial disease; Selective vulnerability.
Figures
References
-
- Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. N Engl J Med. 2001;344:1688–1700. - PubMed
-
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng HX, O’Regan JP, Deng HX, Rahmani Z, Krizus A, McKenna-Yasek D, Cayabyab A, Gaston SM, Berger R, Tanzi RE, Halperin JJ, Herzfeldt B, Van den Bergh R, Hung WY, Bird T, Deng G, Mulder DW, Smyth C, Laing NG, Soriano E, Pericak-Vance MA, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362(6415):59–62. - PubMed
-
- Kwiatkowski TJ, Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, Cortelli P, de Jong PJ, Yoshinaga Y, Haines JL, Pericak-Vance MA, Yan J, Ticozzi N, Siddique T, McKenna-Yasek D, Sapp PC, Horvitz HR, Landers JE, Brown RH., Jr Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323(5918):1205–1208. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous