

Disease mechanisms and therapeutic approaches in spinal muscular atrophy
- PMID: 26063904
- PMCID: PMC4461682
- DOI: 10.1523/JNEUROSCI.0417-15.2015
Disease mechanisms and therapeutic approaches in spinal muscular atrophy
Abstract
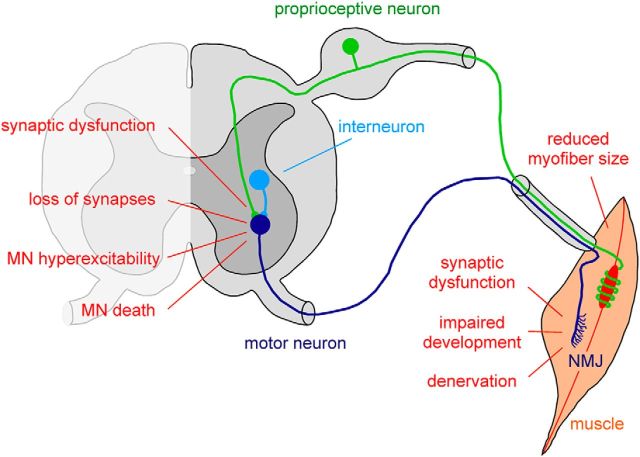
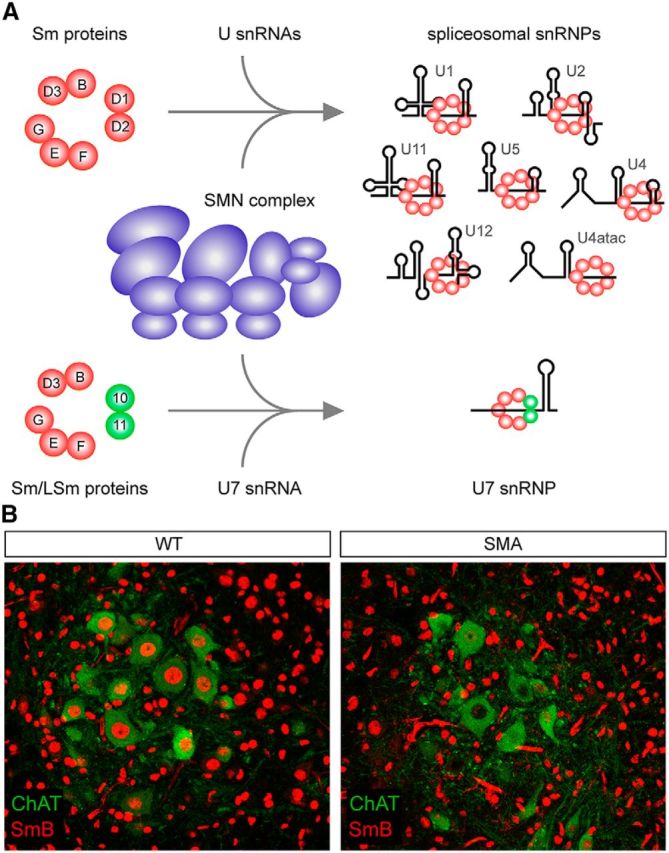
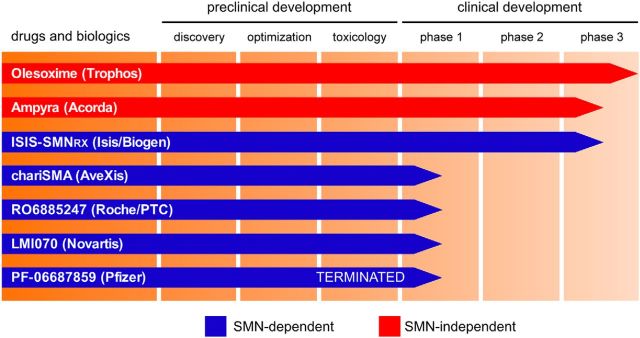
Motor neuron diseases are neurological disorders characterized primarily by the degeneration of spinal motor neurons, skeletal muscle atrophy, and debilitating and often fatal motor dysfunction. Spinal muscular atrophy (SMA) is an autosomal-recessive motor neuron disease of high incidence and severity and the most common genetic cause of infant mortality. SMA is caused by homozygous mutations in the survival motor neuron 1 (SMN1) gene and retention of at least one copy of the hypomorphic gene paralog SMN2. Early studies established a loss-of-function disease mechanism involving ubiquitous SMN deficiency and suggested SMN upregulation as a possible therapeutic approach. In recent years, greater knowledge of the central role of SMN in RNA processing combined with deep characterization of animal models of SMA has significantly advanced our understanding of the cellular and molecular basis of the disease. SMA is emerging as an RNA disease not limited to motor neurons, but one that involves dysfunction of motor circuits that comprise multiple neuronal subpopulations and possibly other cell types. Advances in SMA research have also led to the development of several potential therapeutics shown to be effective in animal models of SMA that are now in clinical trials. These agents offer unprecedented promise for the treatment of this still incurable neurodegenerative disease.
Copyright © 2015 the authors 0270-6474/15/358691-10$15.00/0.
Figures
References
-
- Ackermann B, Kröber S, Torres-Benito L, Borgmann A, Peters M, Hosseini Barkooie SM, Tejero R, Jakubik M, Schreml J, Milbradt J, Wunderlich TF, Riessland M, Tabares L, Wirth B. Plastin 3 ameliorates spinal muscular atrophy via delayed axon pruning and improves neuromuscular junction functionality. Hum Mol Genet. 2013;22:1328–1347. doi: 10.1093/hmg/dds540. - DOI - PubMed
-
- Akten B, Kye MJ, Hao le T, Wertz MH, Singh S, Nie D, Huang J, Merianda TT, Twiss JL, Beattie CE, Steen JA, Sahin M. Interaction of survival of motor neuron (SMN) and HuD proteins with mRNA cpg15 rescues motor neuron axonal deficits. Proc Natl Acad Sci U S A. 2011;108:10337–10342. doi: 10.1073/pnas.1104928108. - DOI - PMC - PubMed
-
- Avila AM, Burnett BG, Taye AA, Gabanella F, Knight MA, Hartenstein P, Cizman Z, Di Prospero NA, Pellizzoni L, Fischbeck KH, Sumner CJ. Trichostatin A increases SMN expression and survival in a mouse model of spinal muscular atrophy. J Clin Invest. 2007;117:659–671. doi: 10.1172/JCI29562. - DOI - PMC - PubMed
-
- Bevan AK, Hutchinson KR, Foust KD, Braun L, McGovern VL, Schmelzer L, Ward JG, Petruska JC, Lucchesi PA, Burghes AH, Kaspar BK. Early heart failure in the SMNDelta7 model of spinal muscular atrophy and correction by postnatal scAAV9-SMN delivery. Hum Mol Genet. 2010;19:3895–3905. doi: 10.1093/hmg/ddq300. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical