

Aquaporin-2: new mutations responsible for autosomal-recessive nephrogenic diabetes insipidus-update and epidemiology
- PMID: 26069764
- PMCID: PMC4400507
- DOI: 10.1093/ckj/sfs029
Aquaporin-2: new mutations responsible for autosomal-recessive nephrogenic diabetes insipidus-update and epidemiology
Abstract
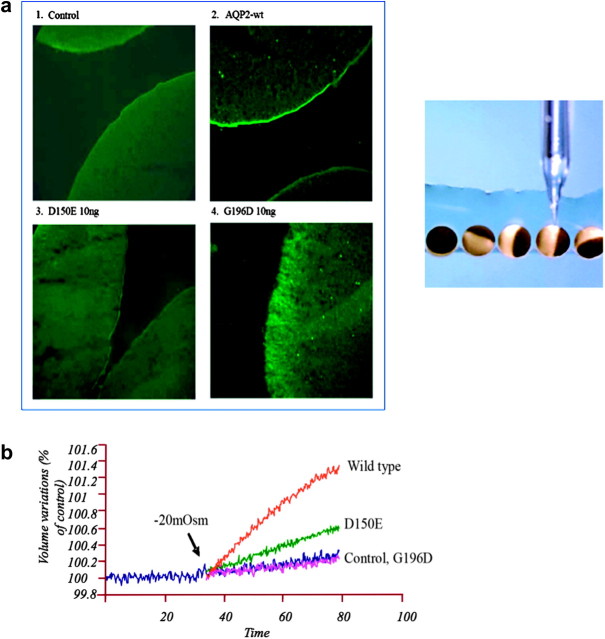
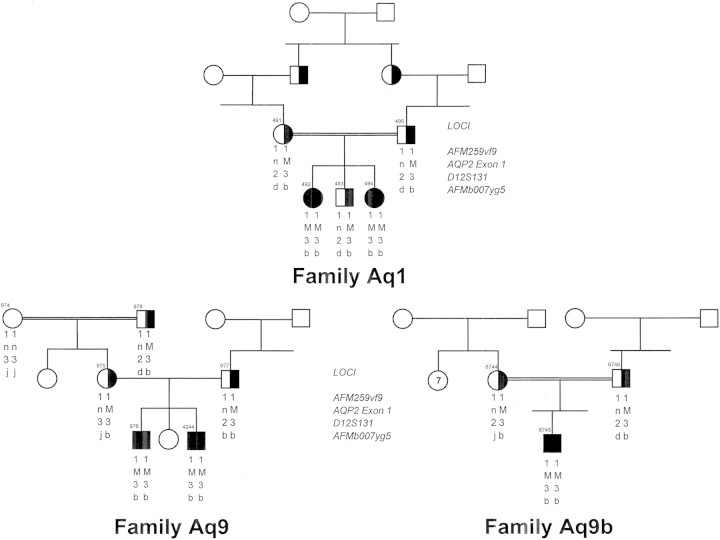
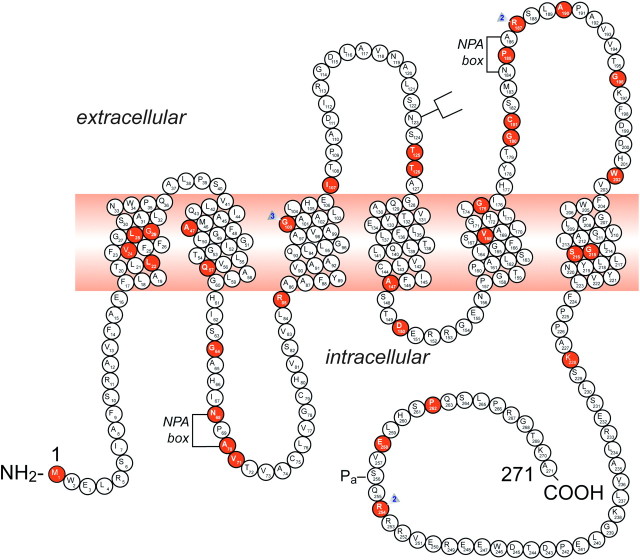
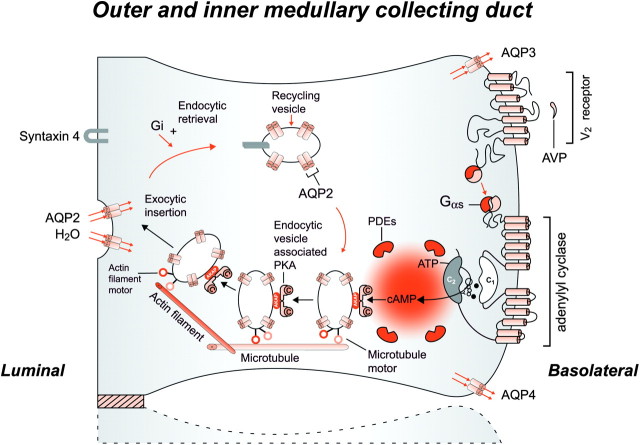
It is clinically useful to distinguish between two types of hereditary nephrogenic diabetes insipidus (NDI): a 'pure' type characterized by loss of water only and a complex type characterized by loss of water and ions. Patients with congenital NDI bearing mutations in the vasopressin 2 receptor gene, AVPR2, or in the aquaporin-2 gene, AQP2, have a pure NDI phenotype with loss of water but normal conservation of sodium, potassium, chloride and calcium. Patients with hereditary hypokalemic salt-losing tubulopathies have a complex phenotype with loss of water and ions. They have polyhydramnios, hypercalciuria and hypo- or isosthenuria and were found to bear KCNJ1 (ROMK) and SLC12A1 (NKCC2) mutations. Patients with polyhydramnios, profound polyuria, hyponatremia, hypochloremia, metabolic alkalosis and sensorineural deafness were found to bear BSND mutations. These clinical phenotypes demonstrate the critical importance of the proteins ROMK, NKCC2 and Barttin to transfer NaCl in the medullary interstitium and thereby to generate, together with urea, a hypertonic milieu. This editorial describes two new developments: (i) the genomic information provided by the sequencing of the AQP2 gene is key to the routine care of these patients, and, as in other genetic diseases, reduces health costs and provides psychological benefits to patients and families and (ii) the expression of AQP2 mutants in Xenopus oocytes and in polarized renal tubular cells recapitulates the clinical phenotypes and reveals a continuum from severe loss of function with urinary osmolalities <150 mOsm/kg H2O to milder defects with urine osmolalities >200 mOsm/kg H2O.
Keywords: aquaporin-2 mutations; autosomal-dominant and -recessive nephrogenic diabetes insipidus; genetic testing; hereditary polyuric states.
Figures

References
-
- Estevez R, Boettger T, Stein V, et al. Barttin is a Cl- channel beta-subunit crucial for renal Cl- reabsorption and inner ear K+ secretion. Nature. 2001;414:558–561. - PubMed
-
- Waldegger S, Jeck N, Barth P, et al. Barttin increases surface expression and changes current properties of ClC-K channels. Pflugers Arch. 2002;444:411–418. - PubMed
-
- Jeck N, Reinalter SC, Henne T, et al. Hypokalemic salt-losing tubulopathy with chronic renal failure and sensorineural deafness. Pediatrics. 2001;108:E5. - PubMed
-
- Birkenhager R, Otto E, Schurmann MJ, et al. Mutation of BSND causes Bartter syndrome with sensorineural deafness and kidney failure. Nat Genet. 2001;29:310–314. - PubMed
-
- Bettinelli A, Bianchetti MG, Girardin E, et al. Use of calcium excretion values to distinguish two forms of primary renal tubular hypokalemic alkalosis: Bartter and Gitelman syndromes. J Pediatr. 1992;120:38–43. - PubMed
LinkOut - more resources
Full Text Sources
