

Recessive nephrocerebellar syndrome on the Galloway-Mowat syndrome spectrum is caused by homozygous protein-truncating mutations of WDR73
- PMID: 26070982
- PMCID: PMC4511861
- DOI: 10.1093/brain/awv153
Recessive nephrocerebellar syndrome on the Galloway-Mowat syndrome spectrum is caused by homozygous protein-truncating mutations of WDR73
Abstract
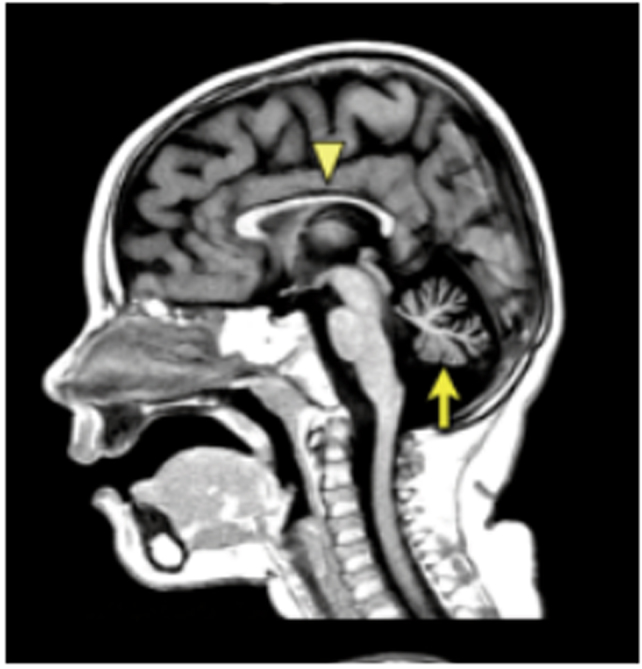
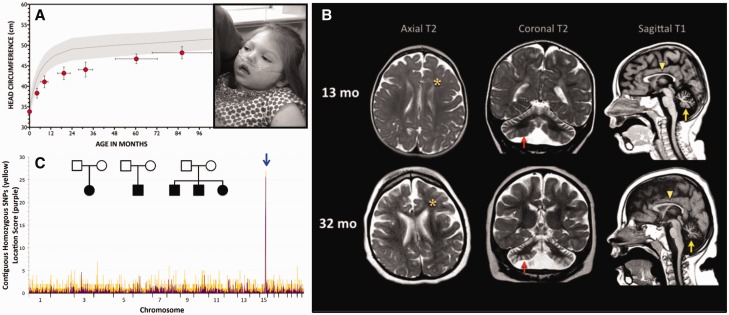
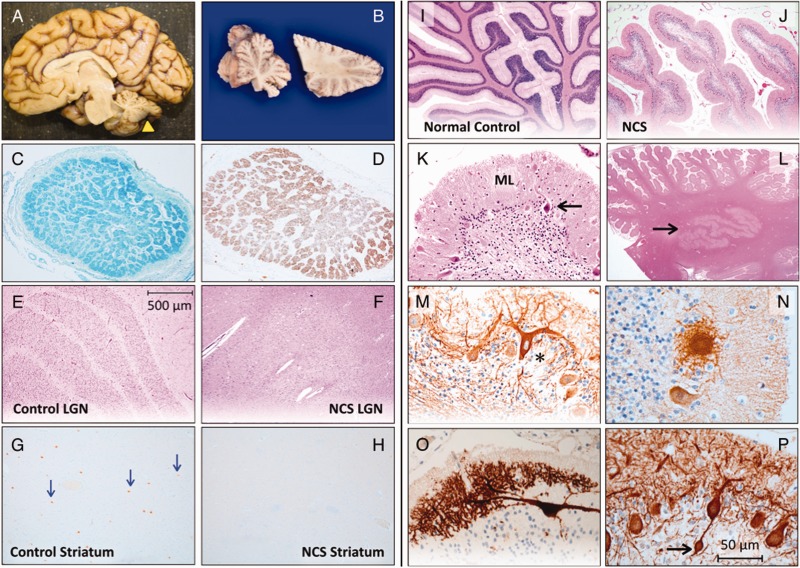
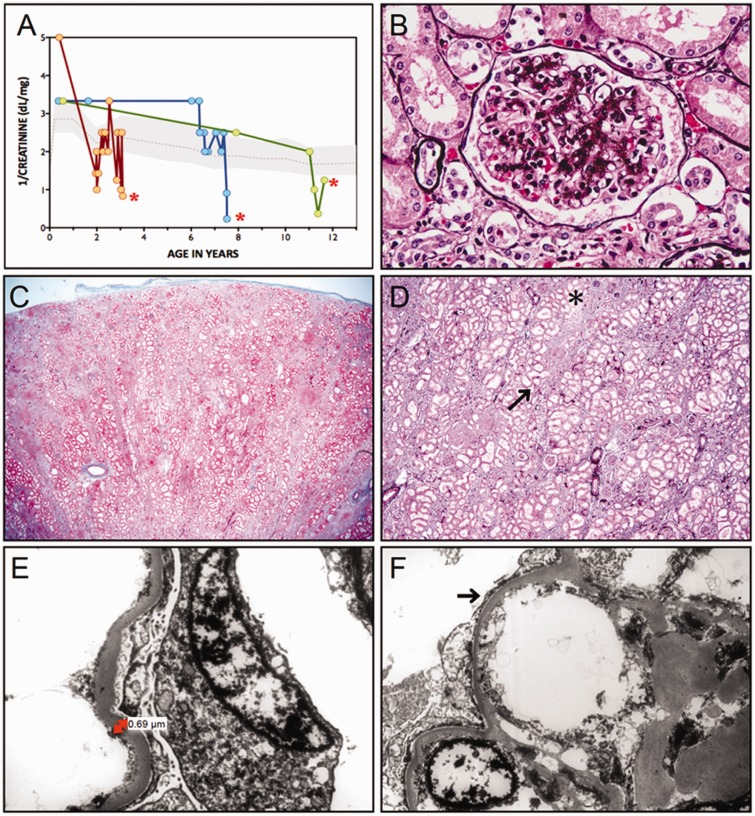
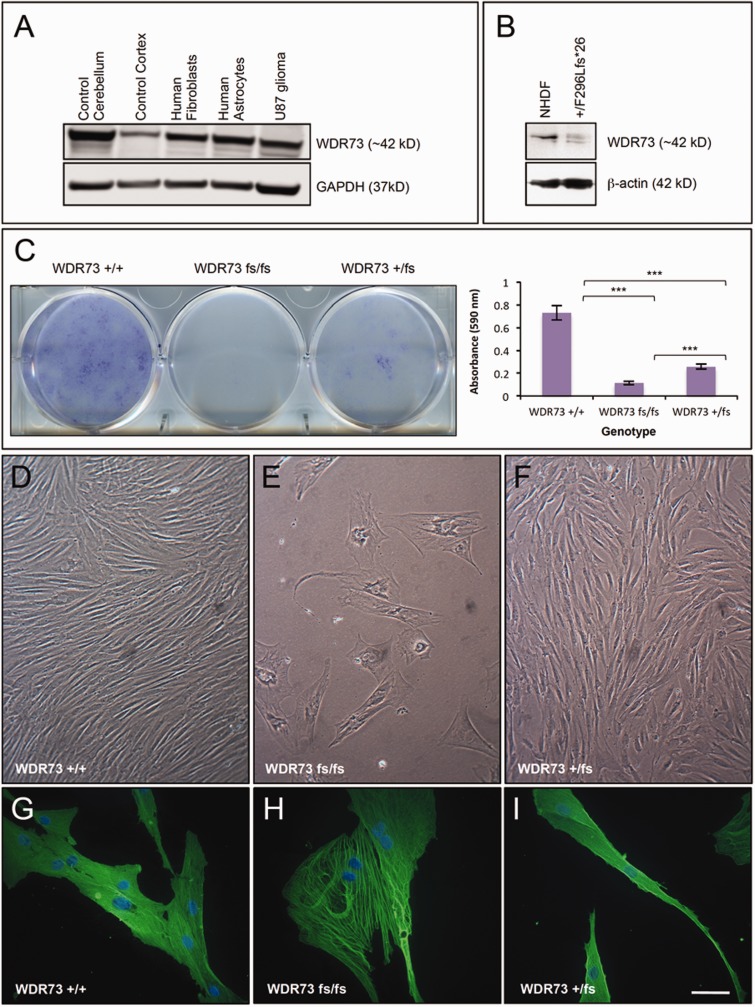
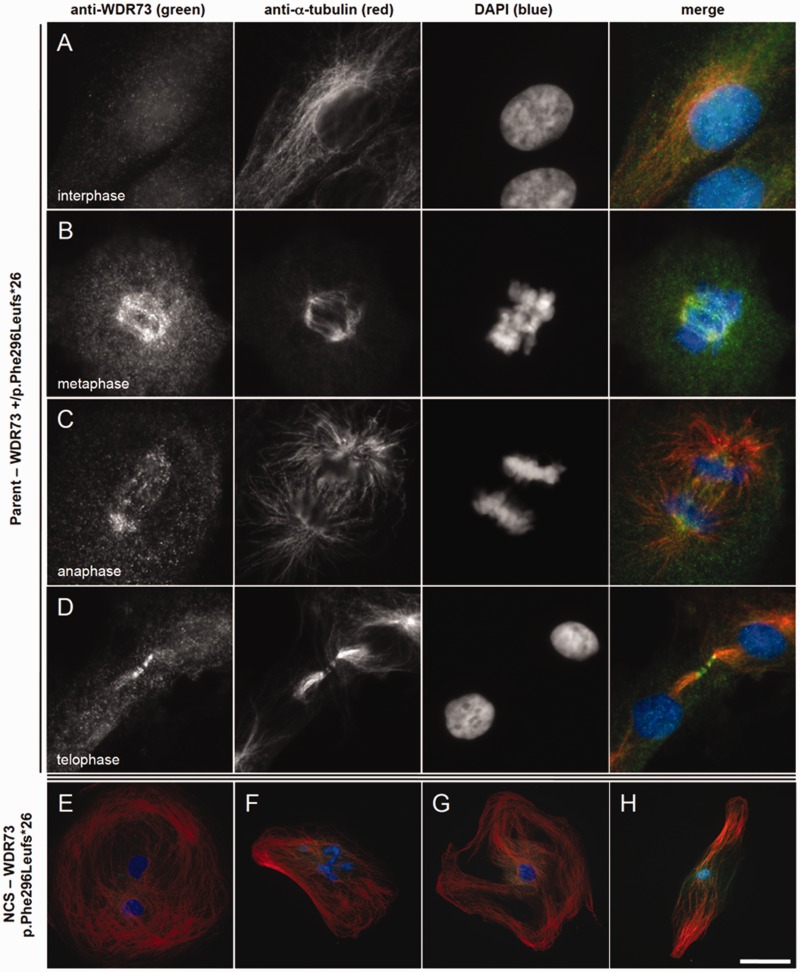
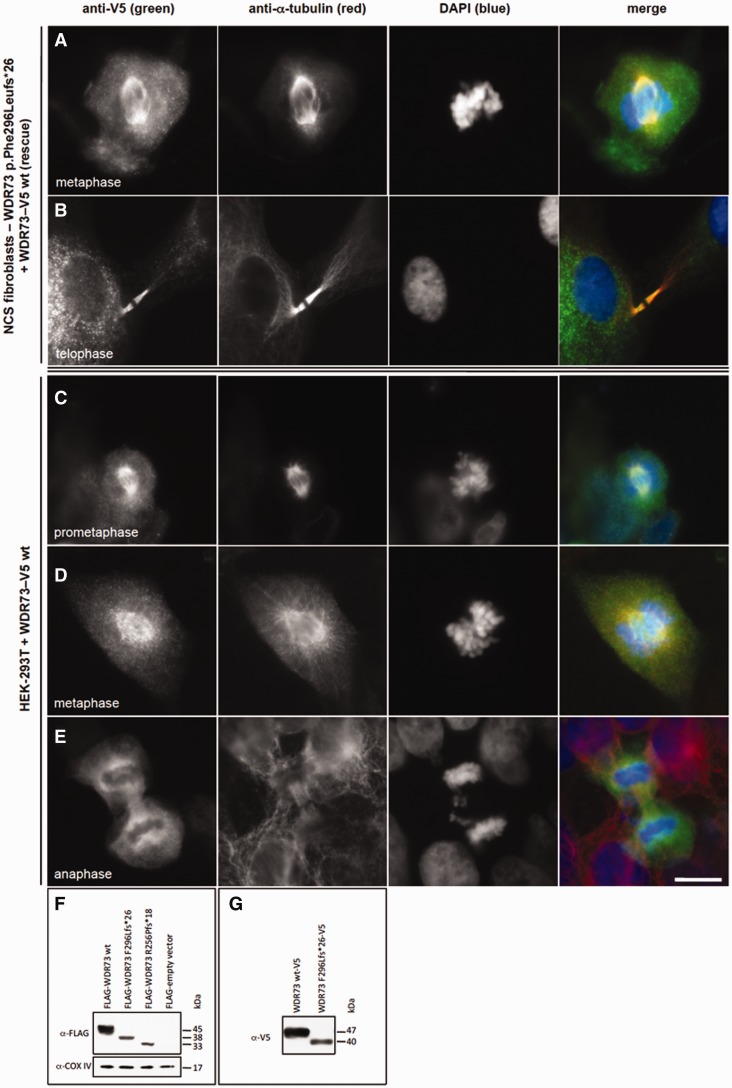
We describe a novel nephrocerebellar syndrome on the Galloway-Mowat syndrome spectrum among 30 children (ages 1.0 to 28 years) from diverse Amish demes. Children with nephrocerebellar syndrome had progressive microcephaly, visual impairment, stagnant psychomotor development, abnormal extrapyramidal movements and nephrosis. Fourteen died between ages 2.7 and 28 years, typically from renal failure. Post-mortem studies revealed (i) micrencephaly without polymicrogyria or heterotopia; (ii) atrophic cerebellar hemispheres with stunted folia, profound granule cell depletion, Bergmann gliosis, and signs of Purkinje cell deafferentation; (iii) selective striatal cholinergic interneuron loss; and (iv) optic atrophy with delamination of the lateral geniculate nuclei. Renal tissue showed focal and segmental glomerulosclerosis and extensive effacement and microvillus transformation of podocyte foot processes. Nephrocerebellar syndrome mapped to 700 kb on chromosome 15, which contained a single novel homozygous frameshift variant (WDR73 c.888delT; p.Phe296Leufs*26). WDR73 protein is expressed in human cerebral cortex, hippocampus, and cultured embryonic kidney cells. It is concentrated at mitotic microtubules and interacts with α-, β-, and γ-tubulin, heat shock proteins 70 and 90 (HSP-70; HSP-90), and the carbamoyl phosphate synthetase 2/aspartate transcarbamylase/dihydroorotase multi-enzyme complex. Recombinant WDR73 p.Phe296Leufs*26 and p.Arg256Profs*18 proteins are truncated, unstable, and show increased interaction with α- and β-tubulin and HSP-70/HSP-90. Fibroblasts from patients homozygous for WDR73 p.Phe296Leufs*26 proliferate poorly in primary culture and senesce early. Our data suggest that in humans, WDR73 interacts with mitotic microtubules to regulate cell cycle progression, proliferation and survival in brain and kidney. We extend the Galloway-Mowat syndrome spectrum with the first description of diencephalic and striatal neuropathology.
Keywords: cerebellar hypoplasia; mTOR; mitosis; nephrosis; progressive microcephaly.
© The Author (2015). Published by Oxford University Press on behalf of the Guarantors of Brain. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures

References
-
- Abraham H, Tornoczky T, Kosztolanyi G, Seress L. Cell formation in the cortical layers of the developing human cerebellum. Int J Dev Neurosci 2001; 19: 53–18. - PubMed
-
- Agromayor M, Martin-Serrano J. Knowing when to cut and run: mechanisms that control cytokinetic abscission. Trends Cell Biol 2013; 23: 433–41. - PubMed
-
- Aosaki T, Miura M, Suzuki T, Nishimura K, Masuda M. Acetylcholine-dopamine balance hypothesis in the striatum: an update. Geriatr Gerontol Int 2010; 10 (Suppl 1): S148–57. - PubMed
-
- Barisoni L. Podocyte biology in segmental sclerosis and progressive glomerular injury. Adv Chronic Kidney Dis 2012; 19: 76–83. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
