

Reconstructing 16S rRNA genes in metagenomic data
- PMID: 26072503
- PMCID: PMC4765874
- DOI: 10.1093/bioinformatics/btv231
Reconstructing 16S rRNA genes in metagenomic data
Abstract
Metagenomic data, which contains sequenced DNA reads of uncultured microbial species from environmental samples, provide a unique opportunity to thoroughly analyze microbial species that have never been identified before. Reconstructing 16S ribosomal RNA, a phylogenetic marker gene, is usually required to analyze the composition of the metagenomic data. However, massive volume of dataset, high sequence similarity between related species, skewed microbial abundance and lack of reference genes make 16S rRNA reconstruction difficult. Generic de novo assembly tools are not optimized for assembling 16S rRNA genes. In this work, we introduce a targeted rRNA assembly tool, REAGO (REconstruct 16S ribosomal RNA Genes from metagenOmic data). It addresses the above challenges by combining secondary structure-aware homology search, zproperties of rRNA genes and de novo assembly. Our experimental results show that our tool can correctly recover more rRNA genes than several popular generic metagenomic assembly tools and specially designed rRNA construction tools.
Availability and implementation: The source code of REAGO is freely available at https://github.com/chengyuan/reago.
© The Author 2015. Published by Oxford University Press.
Figures
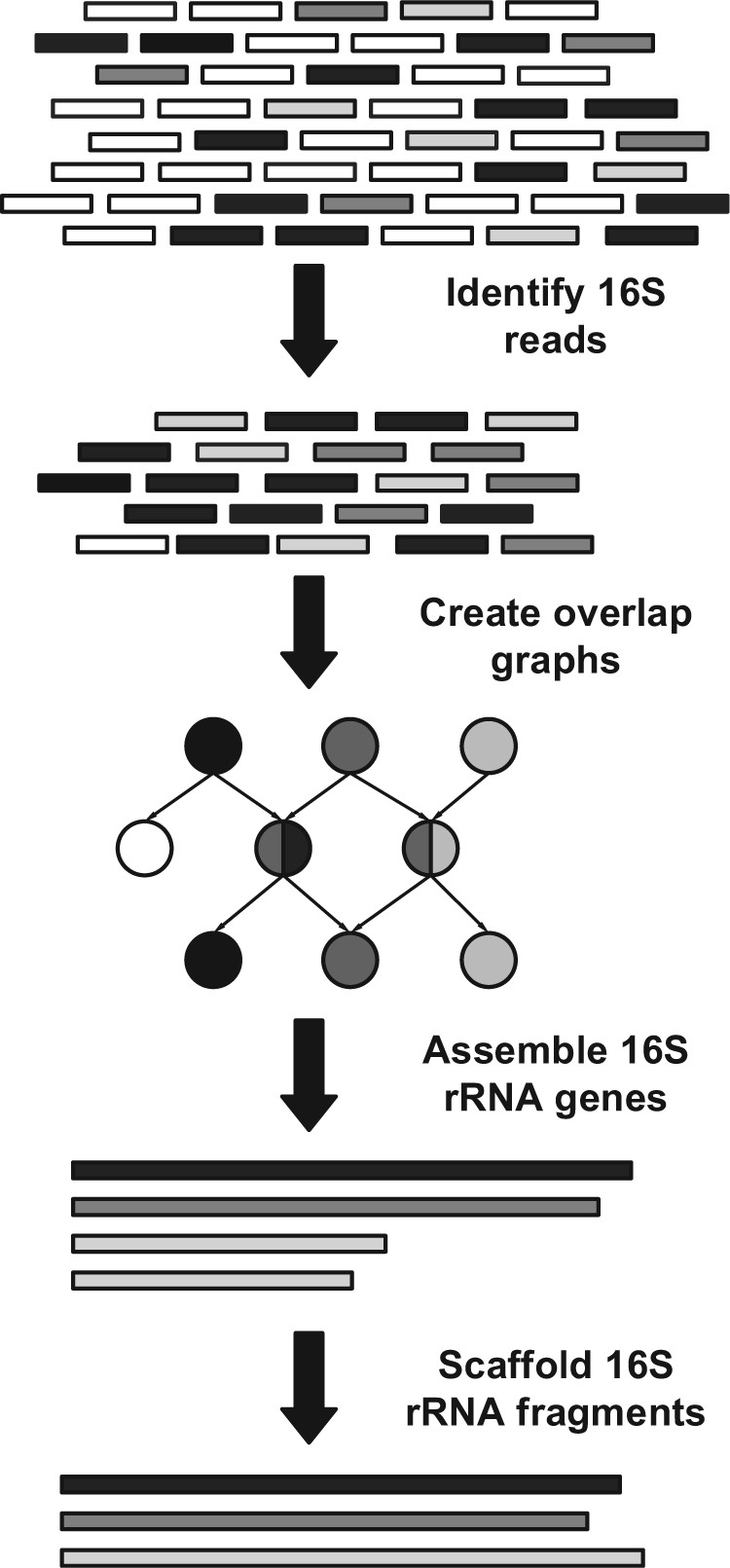
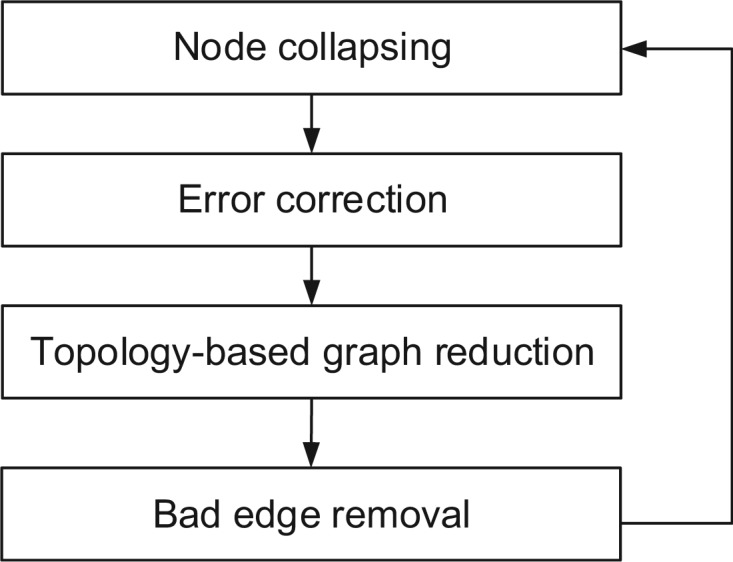
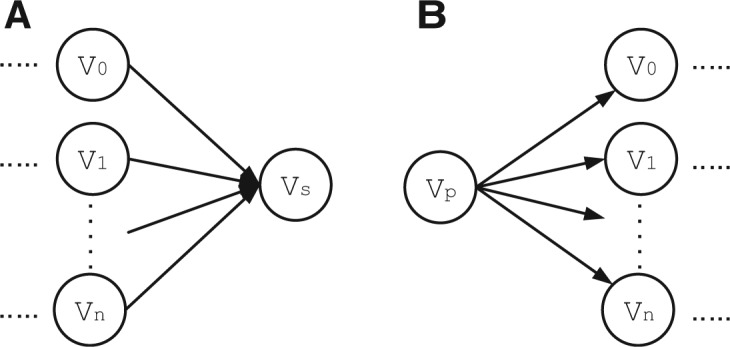
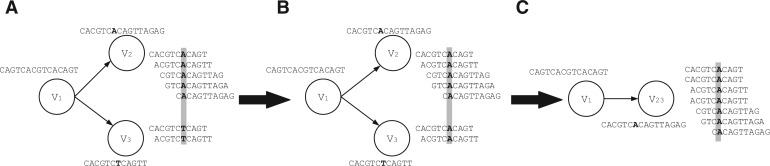

References
-
- Altschul S.F., et al. (1990) Basic local alignment search tool. J. Mol. Biol., 215, 403–410. - PubMed
-
- Berg R.D. (1996) The indigenous gastrointestinal microflora. Trends Microbiol., 4, 430–435. - PubMed
-
- Christen R. (2008) Global sequencing: a review of current molecular data and new methods available to assess microbial diversity. Microbes Environ. JSME, 23, 253–268. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
