

IgRepertoireConstructor: a novel algorithm for antibody repertoire construction and immunoproteogenomics analysis
- PMID: 26072509
- PMCID: PMC4542777
- DOI: 10.1093/bioinformatics/btv238
IgRepertoireConstructor: a novel algorithm for antibody repertoire construction and immunoproteogenomics analysis
Abstract
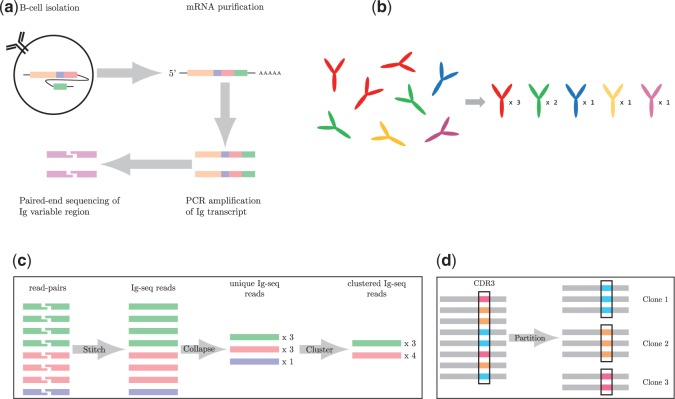
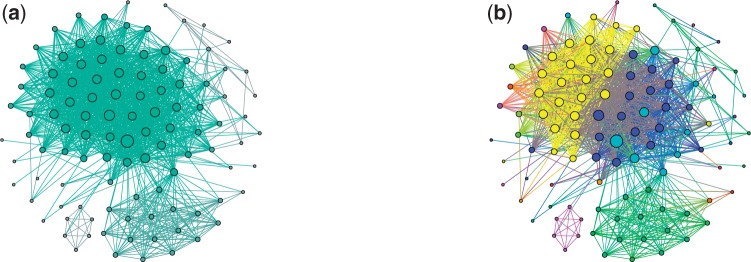
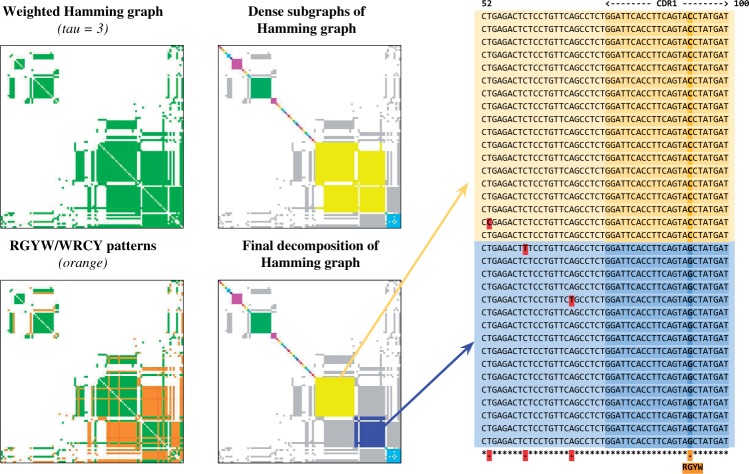
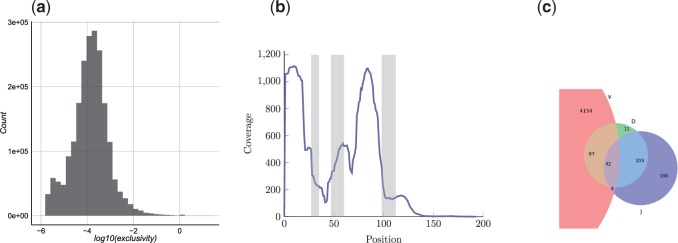
The analysis of concentrations of circulating antibodies in serum (antibody repertoire) is a fundamental, yet poorly studied, problem in immunoinformatics. The two current approaches to the analysis of antibody repertoires [next generation sequencing (NGS) and mass spectrometry (MS)] present difficult computational challenges since antibodies are not directly encoded in the germline but are extensively diversified by somatic recombination and hypermutations. Therefore, the protein database required for the interpretation of spectra from circulating antibodies is custom for each individual. Although such a database can be constructed via NGS, the reads generated by NGS are error-prone and even a single nucleotide error precludes identification of a peptide by the standard proteomics tools. Here, we present the IgRepertoireConstructor algorithm that performs error-correction of immunosequencing reads and uses mass spectra to validate the constructed antibody repertoires.
Availability and implementation: IgRepertoireConstructor is open source and freely available as a C++ and Python program running on all Unix-compatible platforms. The source code is available from http://bioinf.spbau.ru/igtools.
Contact: ppevzner@ucsd.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
© The Author 2015. Published by Oxford University Press.
Figures
References
-
- Ben-Dor A., et al. . (1999) Clustering gene expression patterns. J. Comp. Biol., 6, 281–297. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
