

De novo meta-assembly of ultra-deep sequencing data
- PMID: 26072514
- PMCID: PMC4765875
- DOI: 10.1093/bioinformatics/btv226
De novo meta-assembly of ultra-deep sequencing data
Abstract
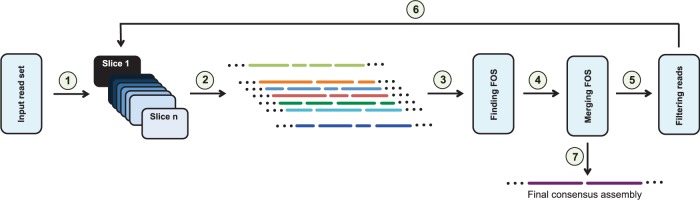
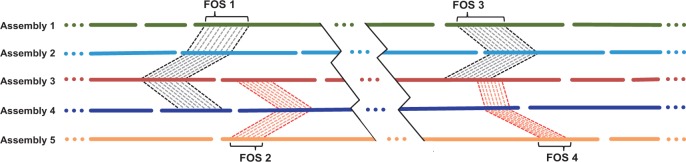
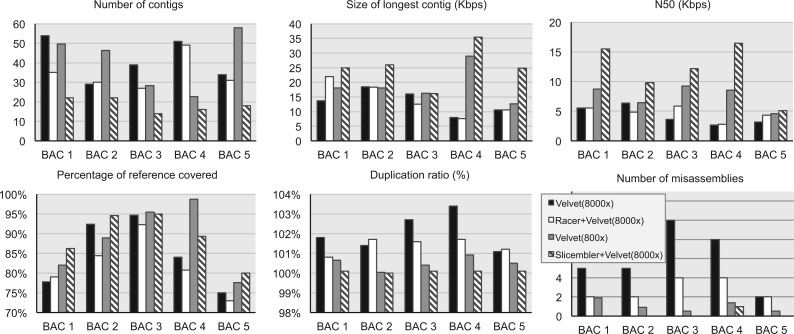
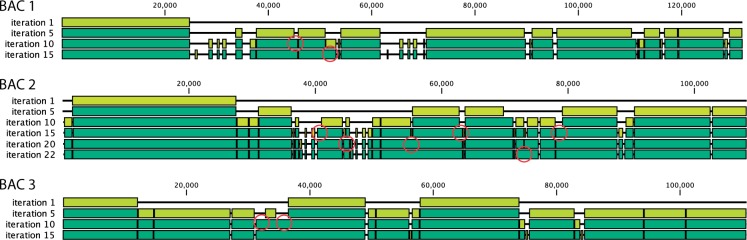
We introduce a new divide and conquer approach to deal with the problem of de novo genome assembly in the presence of ultra-deep sequencing data (i.e. coverage of 1000x or higher). Our proposed meta-assembler Slicembler partitions the input data into optimal-sized 'slices' and uses a standard assembly tool (e.g. Velvet, SPAdes, IDBA_UD and Ray) to assemble each slice individually. Slicembler uses majority voting among the individual assemblies to identify long contigs that can be merged to the consensus assembly. To improve its efficiency, Slicembler uses a generalized suffix tree to identify these frequent contigs (or fraction thereof). Extensive experimental results on real ultra-deep sequencing data (8000x coverage) and simulated data show that Slicembler significantly improves the quality of the assembly compared with the performance of the base assembler. In fact, most of the times, Slicembler generates error-free assemblies. We also show that Slicembler is much more resistant against high sequencing error rate than the base assembler.
Availability and implementation: Slicembler can be accessed at http://slicembler.cs.ucr.edu/.
© The Author 2015. Published by Oxford University Press.
Figures
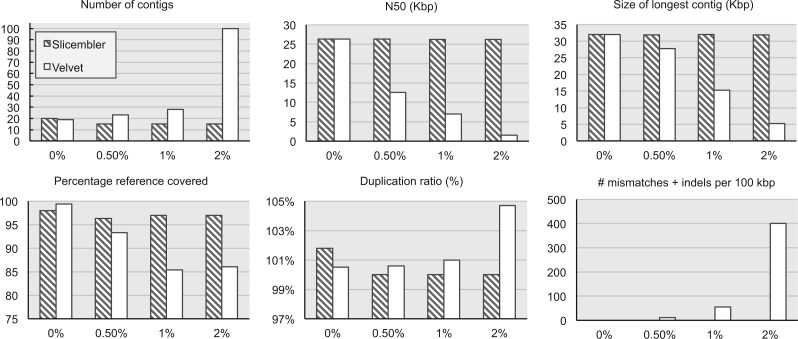
References
-
- Beerenwinkel N., Zagordi O. (2011) Ultra-deep sequencing for the analysis of viral populations. Curr. Opin. Virol., 1, 413–418. - PubMed
-
- Brown C.T., et al. (2012) A reference-free algorithm for computational normalization of shotgun sequencing data. arXiv:1203.4802.
