

2014 Update of the Alzheimer's Disease Neuroimaging Initiative: A review of papers published since its inception
- PMID: 26073027
- PMCID: PMC5469297
- DOI: 10.1016/j.jalz.2014.11.001
2014 Update of the Alzheimer's Disease Neuroimaging Initiative: A review of papers published since its inception
Abstract
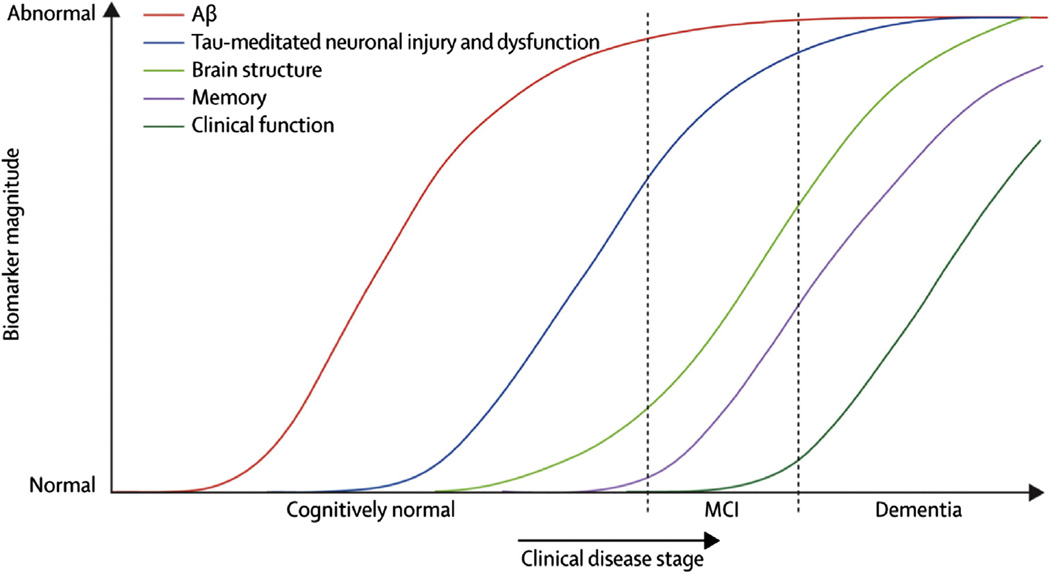
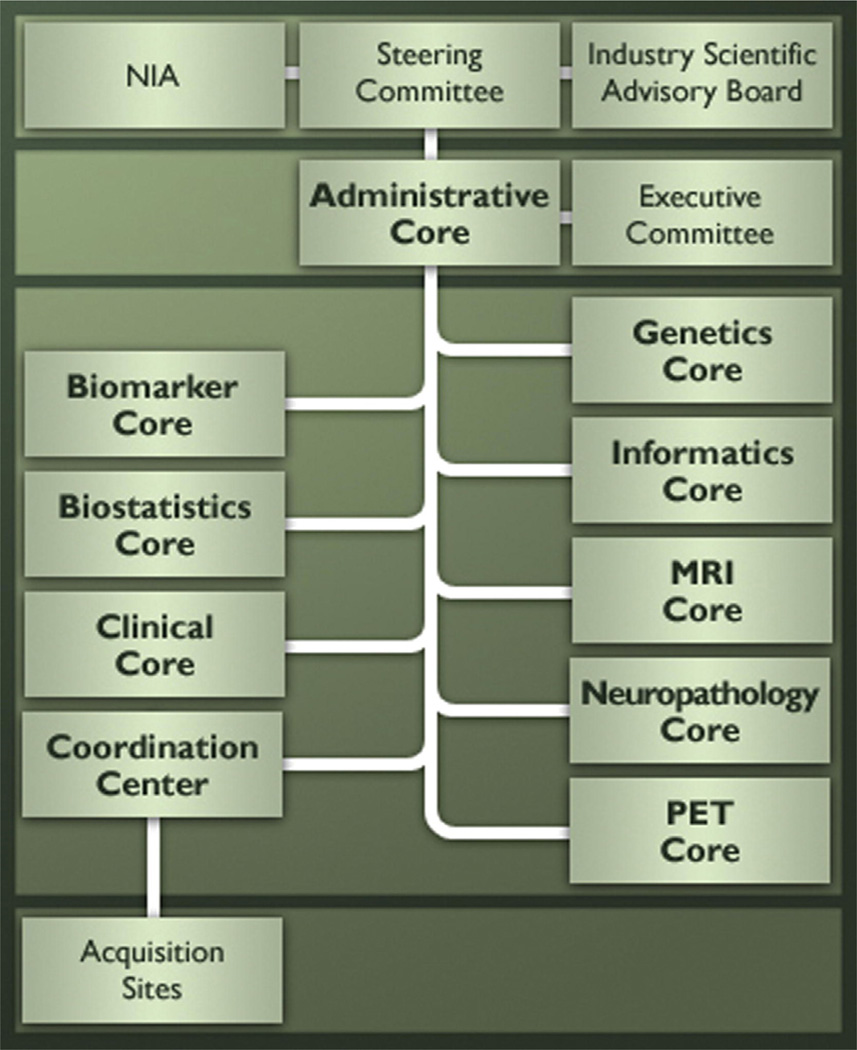
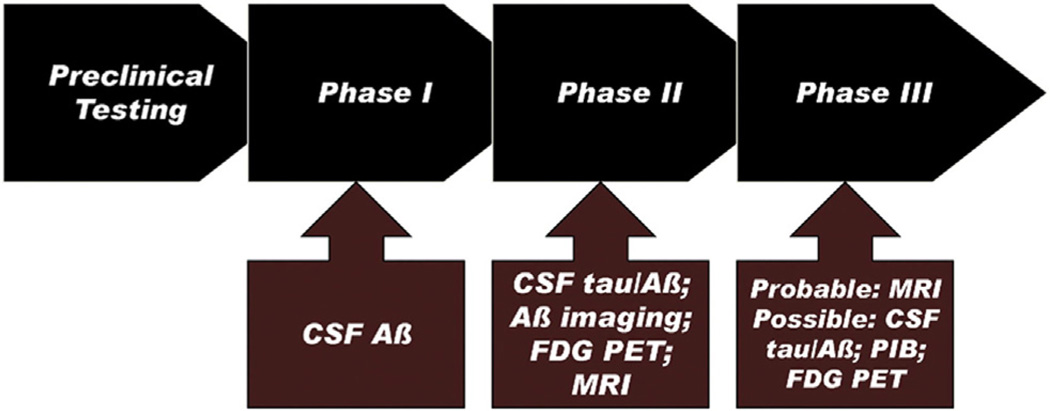
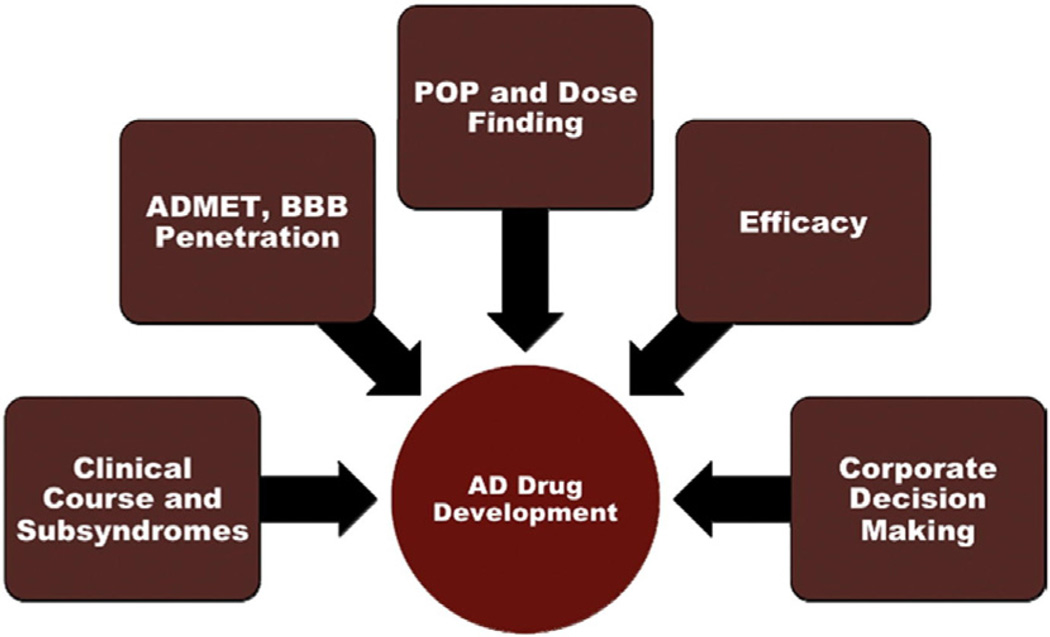
The Alzheimer's Disease Neuroimaging Initiative (ADNI) is an ongoing, longitudinal, multicenter study designed to develop clinical, imaging, genetic, and biochemical biomarkers for the early detection and tracking of Alzheimer's disease (AD). The initial study, ADNI-1, enrolled 400 subjects with early mild cognitive impairment (MCI), 200 with early AD, and 200 cognitively normal elderly controls. ADNI-1 was extended by a 2-year Grand Opportunities grant in 2009 and by a competitive renewal, ADNI-2, which enrolled an additional 550 participants and will run until 2015. This article reviews all papers published since the inception of the initiative and summarizes the results to the end of 2013. The major accomplishments of ADNI have been as follows: (1) the development of standardized methods for clinical tests, magnetic resonance imaging (MRI), positron emission tomography (PET), and cerebrospinal fluid (CSF) biomarkers in a multicenter setting; (2) elucidation of the patterns and rates of change of imaging and CSF biomarker measurements in control subjects, MCI patients, and AD patients. CSF biomarkers are largely consistent with disease trajectories predicted by β-amyloid cascade (Hardy, J Alzheimer's Dis 2006;9(Suppl 3):151-3) and tau-mediated neurodegeneration hypotheses for AD, whereas brain atrophy and hypometabolism levels show predicted patterns but exhibit differing rates of change depending on region and disease severity; (3) the assessment of alternative methods of diagnostic categorization. Currently, the best classifiers select and combine optimum features from multiple modalities, including MRI, [(18)F]-fluorodeoxyglucose-PET, amyloid PET, CSF biomarkers, and clinical tests; (4) the development of blood biomarkers for AD as potentially noninvasive and low-cost alternatives to CSF biomarkers for AD diagnosis and the assessment of α-syn as an additional biomarker; (5) the development of methods for the early detection of AD. CSF biomarkers, β-amyloid 42 and tau, as well as amyloid PET may reflect the earliest steps in AD pathology in mildly symptomatic or even nonsymptomatic subjects and are leading candidates for the detection of AD in its preclinical stages; (6) the improvement of clinical trial efficiency through the identification of subjects most likely to undergo imminent future clinical decline and the use of more sensitive outcome measures to reduce sample sizes. Multimodal methods incorporating APOE status and longitudinal MRI proved most highly predictive of future decline. Refinements of clinical tests used as outcome measures such as clinical dementia rating-sum of boxes further reduced sample sizes; (7) the pioneering of genome-wide association studies that leverage quantitative imaging and biomarker phenotypes, including longitudinal data, to confirm recently identified loci, CR1, CLU, and PICALM and to identify novel AD risk loci; (8) worldwide impact through the establishment of ADNI-like programs in Japan, Australia, Argentina, Taiwan, China, Korea, Europe, and Italy; (9) understanding the biology and pathobiology of normal aging, MCI, and AD through integration of ADNI biomarker and clinical data to stimulate research that will resolve controversies about competing hypotheses on the etiopathogenesis of AD, thereby advancing efforts to find disease-modifying drugs for AD; and (10) the establishment of infrastructure to allow sharing of all raw and processed data without embargo to interested scientific investigators throughout the world.
Keywords: Alzheimer's disease; Amyloid; Biomarker; Mild cognitive impairment; Tau.
Published by Elsevier Inc.
Conflict of interest statement
Dallas P. Veitch has no conflicts to report.
Robert C. Green has no conflicts of interest to report.
Danielle Harvey has no conflicts of interest to report.
Judith A. Siuciak has no conflicts of interest to report.
Arthur W. Toga has no conflicts of interest to report.
Figures




















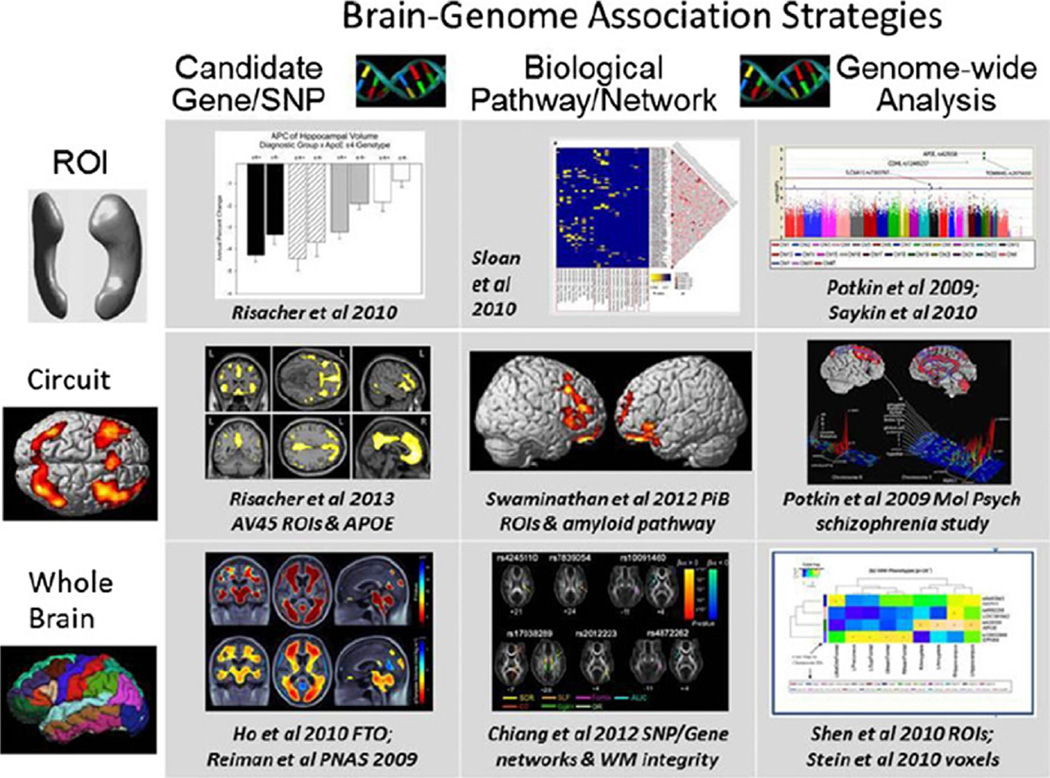
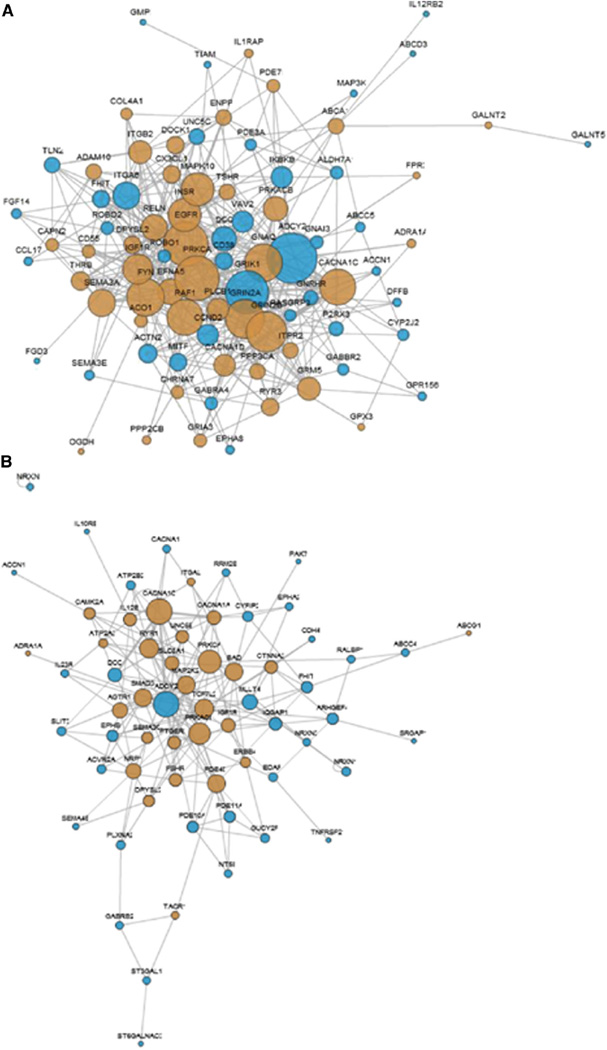
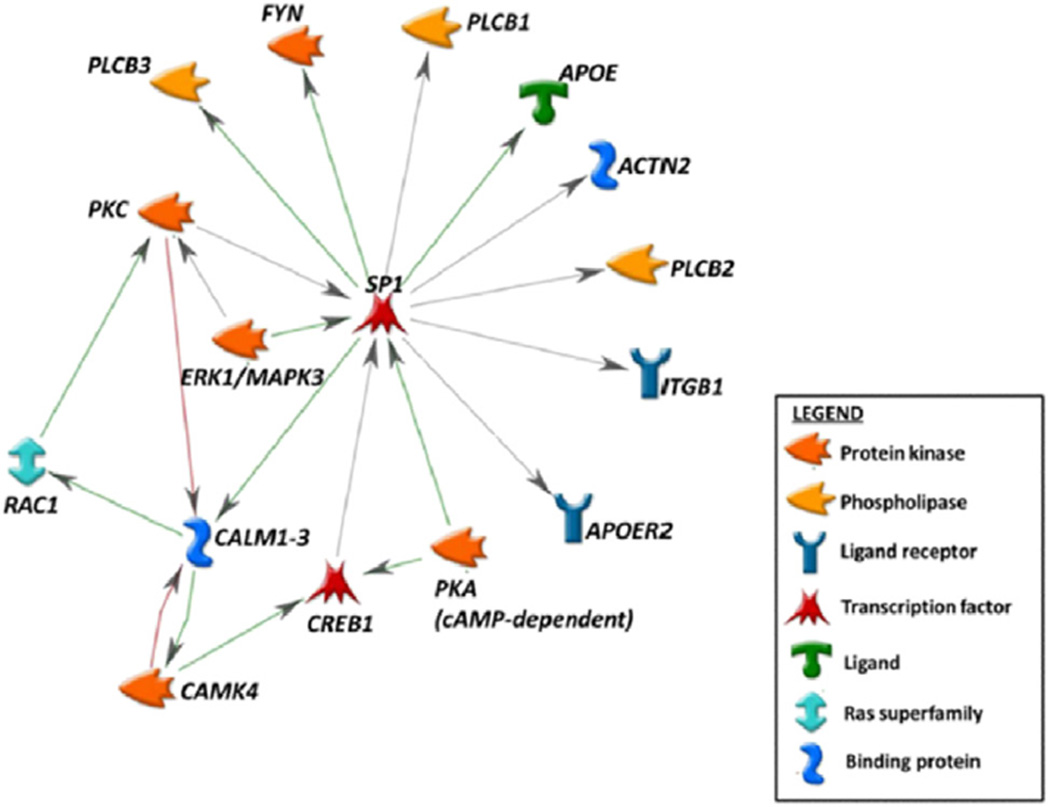
References
Appendix
1 Publications arising from AIBL
1.1 AIBL Publication list, 2009–present
1.1.1 2009
-
- Bourgeat P, Chetelat G, Villemagne VL, Fripp J, Raniga P, Acosta O, et al. B-amyloid burden in the temporal neocortex is related to hippocampal atrophy in elderly subjects without dementia. Neurology. 2010;74:121–127. - PubMed
-
- Ellis KA, Bush AI, Darby D, De Fazio D, Foster J, Hudson P, et al. The Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging: methodology and baseline characteristics of 1112 individuals recruited for a longitudinal study of Alzheimer’s disease. Int Psychogeriatr. 2009;21:672–687. - PubMed
-
- Fodero-Tavoletti MT, Cappai R, McLean CA, Pike KE, Adlard PA, Cowie T, et al. Amyloid imaging in Alzheimer’s disease and other dementias. Brain Imaging Behav. 2009;3:246–261. - PubMed
-
- Fodero-Tavoletti MT, Rowe CC, McLean CA, Leone L, Li QX, Masters CL, Cappai R, Villemagne VL. Characterization of PiB binding to white matter in AD and other dementias. J Nucl Med. 2009;50:198–204. - PubMed
-
- Fodero-Tavoletti MT, Mulligan RS, Okamura N, Furumoto S, Rowe CC, Kudo Y, et al. In vitro characterisation of BF227 binding to α-synuclein/Lewy Bodies. Eur J Pharmacol. 2009;617:54–58. - PubMed
1.1.2 2010
-
- Bourgeat P, Chételat G, Villemagne VL, Fripp J, Raniga P, Pike K, et al. β-amyloid burden in the temporal neocortex is related to hippocampal atrophy in elderly subjects without dementia. Neurology. 2010;74:121–127. - PubMed
-
- Ellis KA, Rowe CC, Villemagne VL, Martins RN, Masters CL, Salvado O, Szoeke C, Ames D the AIBL Research Group. Addressing population aging and Alzheimer’s disease through the Australian imaging biomarkers and lifestyle study: collaboration with the Alzheimer’s Disease Neuroimaging Initiative. Alzheimers Dement. 2010;6:291–296. - PubMed
-
- Chételat G, Villemagne VL, Bourgeat P, Pike KE, Jones G, Ames D, et al. Relationship between atrophy and β-amyloid deposition in Alzheimer disease. Ann Neurol. 2010;67:317–324. [See also editorial: Rabinovici GD, Roberson ED. Beyond diagnosis: what biomarkers are teaching us about the “bio”logy of Alzheimer disease. Ann Neurol 2010;67:283–5.] - PubMed
-
- Villemagne VL, Pike K, Pejoska S, Boyd A, Power M, Jones G, Masters CL, Rowe CC. 11C-PiB PET ABri imaging in Worster-Drought syndrome (Familial British Dementia): a case report. J Alzheimers Dis. 2010;19:423–428. - PubMed
1.1.3 2011
1.1.3.1 Published
-
- Ellis KA, Rowe CC, Szoeke C, Villemagne VL, Ames D, Chételat G, et al. Advances in structural and molecular neuroimaging in Alzheimer’s disease. Med J Aust. 2011;194:S20–S23. - PubMed
-
- Bahar-Fuchs A, Moss S, Pike KE, Villemagne VL, Masters CL, Rowe C, Savage G. Olfactory deficits and Aβ burden in AD, MCI and healthy ageing: a PiB PET Study. J Alzheimers Dis. 2010;22:1081–1087. - PubMed
-
- Gupta VB, Laws SM, Villemagne VL, Ames D, Bush AI, Ellis KA, et al. Plasma Apolipoprotein E and Alzheimer’s disease risk: the AIBL study of ageing. Neurology. 2011;76:1091–1098. - PubMed
-
- Sittironnait G, Ames D, Bush AI, Faux N, Flicker L, Foster J, et al. Effects of anticholinergic drugs on cognitive function in older Australians: results from the AIBL Study. Dement Geriatr Cogn Disord (Special ASIA issue) 2011;31:173–178. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- P01 AG009215/AG/NIA NIH HHS/United States
- P30 AG010133/AG/NIA NIH HHS/United States
- P01 AG017586/AG/NIA NIH HHS/United States
- P50 NS053488/NS/NINDS NIH HHS/United States
- RC2 AG036535/AG/NIA NIH HHS/United States
- P01 AG019724/AG/NIA NIH HHS/United States
- U01 AG010483/AG/NIA NIH HHS/United States
- P30 AG010129/AG/NIA NIH HHS/United States
- P30 CA093373/CA/NCI NIH HHS/United States
- P30 AG010124/AG/NIA NIH HHS/United States
- R01 GM088336/GM/NIGMS NIH HHS/United States
- R01 AG019771/AG/NIA NIH HHS/United States
- U01 AG024904/AG/NIA NIH HHS/United States
- R01 AG016381/AG/NIA NIH HHS/United States
- R25 RR026008/RR/NCRR NIH HHS/United States
- P01 AG003991/AG/NIA NIH HHS/United States
- P50 AG005681/AG/NIA NIH HHS/United States
- R01 AG012975/AG/NIA NIH HHS/United States
- U01 AG029213/AG/NIA NIH HHS/United States
- UL1 RR024146/RR/NCRR NIH HHS/United States
- P30 AG036468/AG/NIA NIH HHS/United States
- R01 AG030048/AG/NIA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
