

High-resolution profiling of histone h3 lysine 36 trimethylation in metastatic renal cell carcinoma
- PMID: 26073078
- PMCID: PMC4679725
- DOI: 10.1038/onc.2015.221
High-resolution profiling of histone h3 lysine 36 trimethylation in metastatic renal cell carcinoma
Abstract
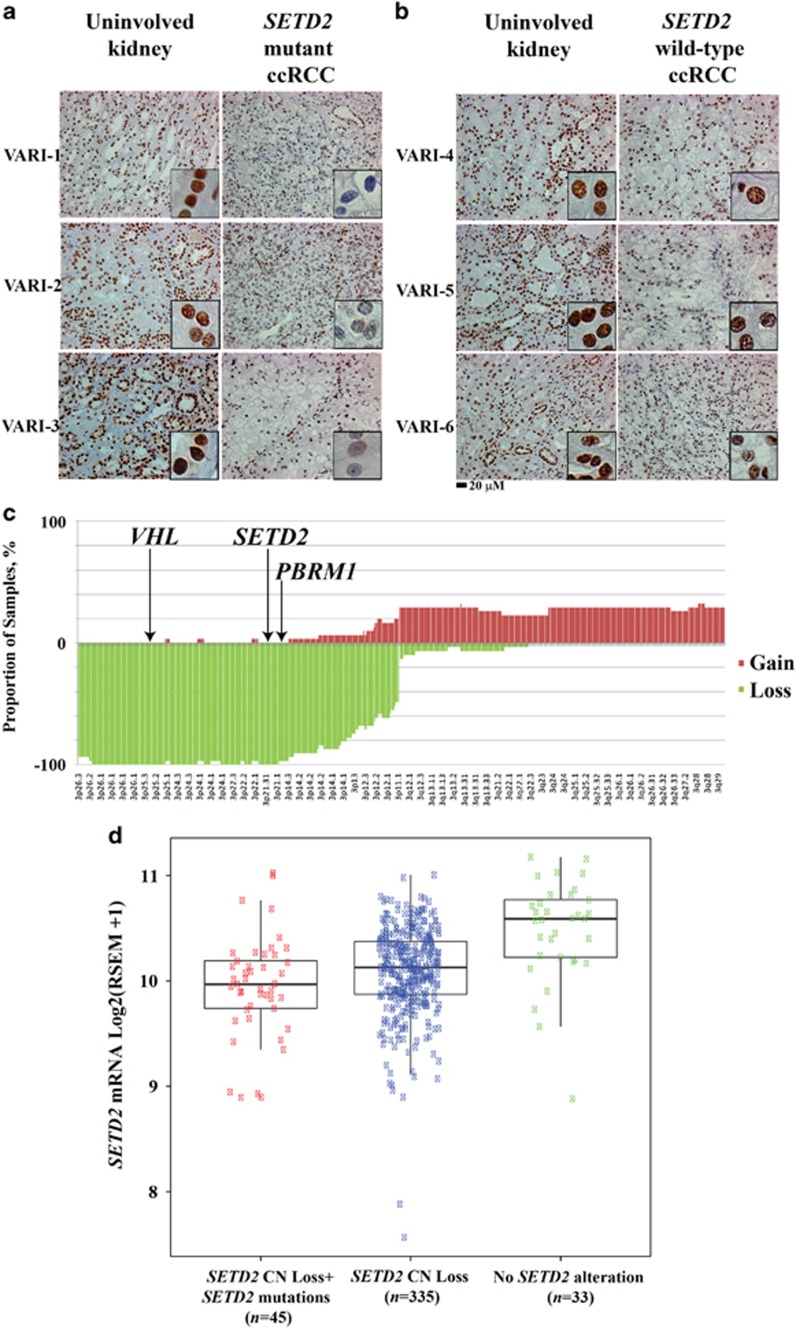
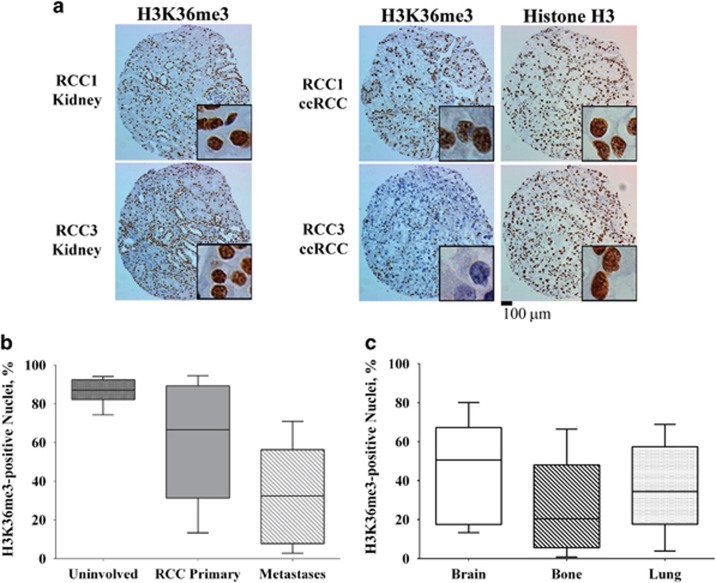
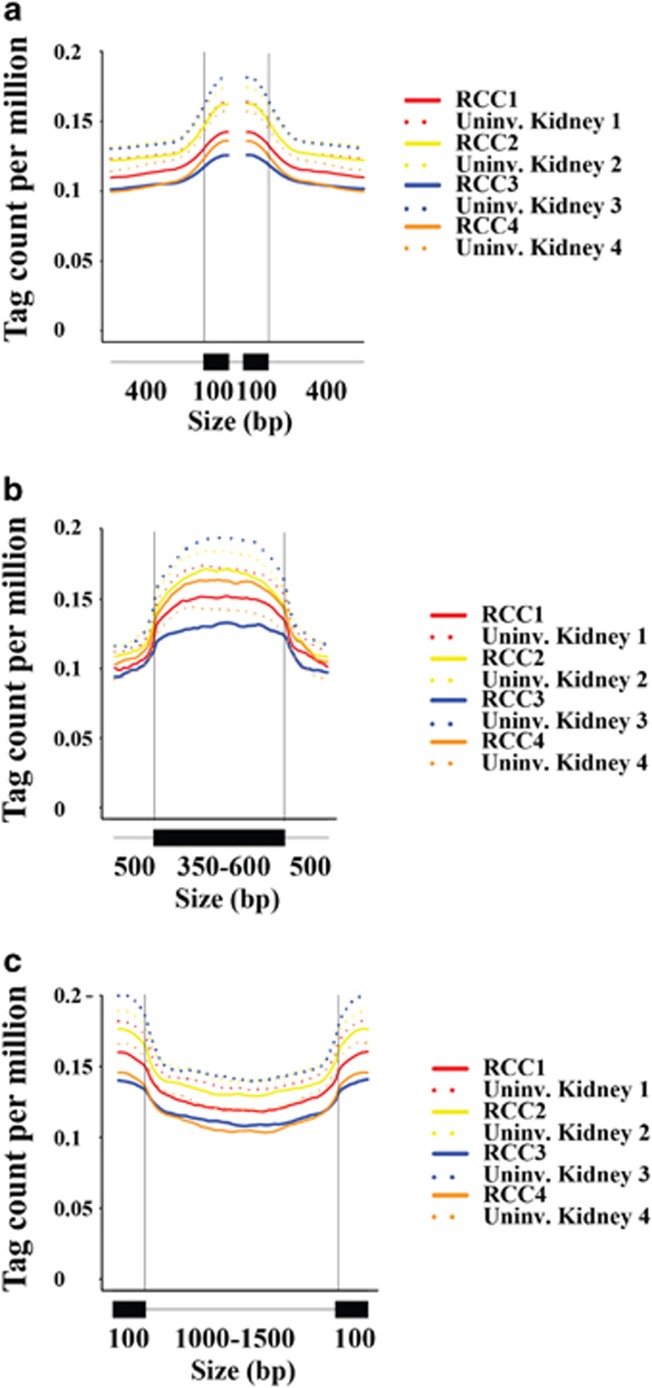
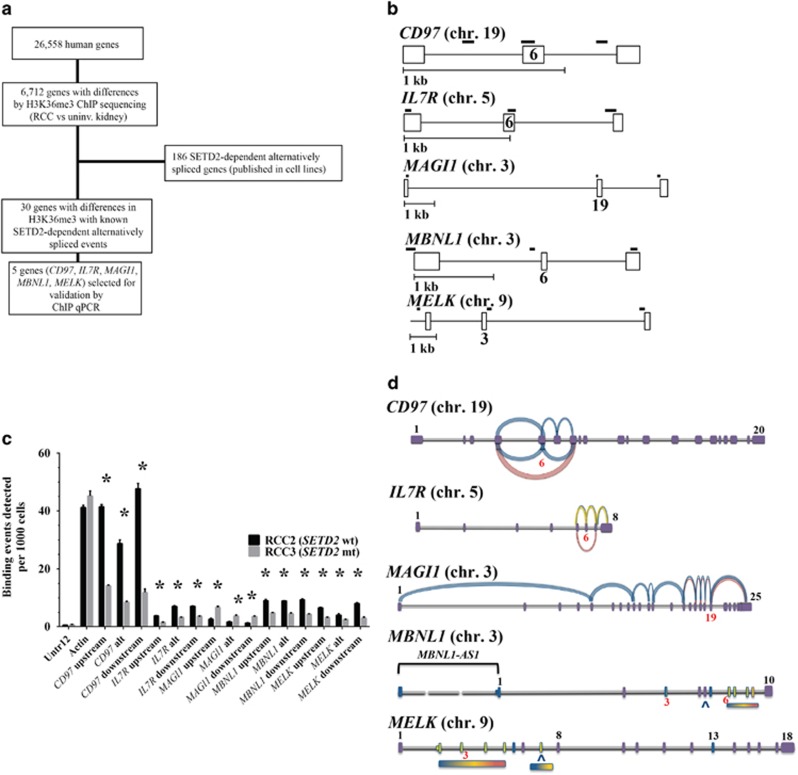
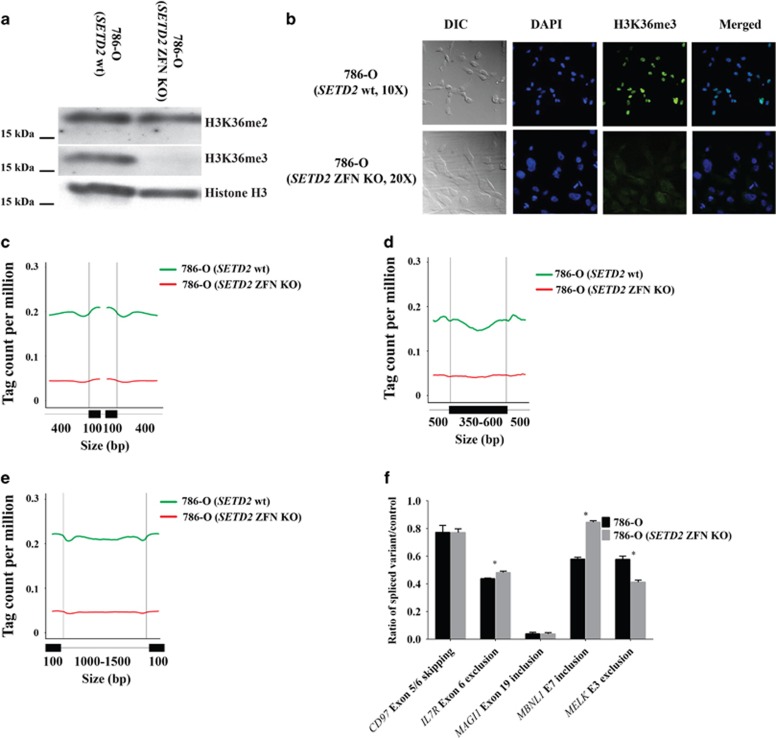
Mutations in SETD2, a histone H3 lysine trimethyltransferase, have been identified in clear cell renal cell carcinoma (ccRCC); however it is unclear if loss of SETD2 function alters the genomic distribution of histone 3 lysine 36 trimethylation (H3K36me3) in ccRCC. Furthermore, published epigenomic profiles are not specific to H3K36me3 or metastatic tumors. To determine if progressive SETD2 and H3K36me3 dysregulation occurs in metastatic tumors, H3K36me3, SETD2 copy number (CN) or SETD2 mRNA abundance was assessed in two independent cohorts: metastatic ccRCC (n=71) and the Cancer Genome Atlas Kidney Renal Clear Cell Carcinoma data set (n=413). Although SETD2 CN loss occurs with high frequency (>90%), H3K36me3 is not significantly impacted by monoallelic loss of SETD2. H3K36me3-positive nuclei were reduced an average of ~20% in primary ccRCC (90% positive nuclei in uninvolved vs 70% positive nuclei in ccRCC) and reduced by ~60% in metastases (90% positive in uninvolved kidney vs 30% positive in metastases) (P<0.001). To define a kidney-specific H3K36me3 profile, we generated genome-wide H3K36me3 profiles from four cytoreductive nephrectomies and SETD2 isogenic renal cell carcinoma (RCC) cell lines using chromatin immunoprecipitation coupled with high-throughput DNA sequencing and RNA sequencing. SETD2 loss of methyltransferase activity leads to regional alterations of H3K36me3 associated with aberrant RNA splicing in a SETD2 mutant RCC and SETD2 knockout cell line. These data suggest that during progression of ccRCC, a decline in H3K36me3 is observed in distant metastases, and regional H3K36me3 alterations influence alternative splicing in ccRCC.
Figures
References
-
- Cohen HT, McGovern FJ. Renal-cell carcinoma. N Engl J Med 2005; 353: 2477–2490. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
