

MemProtMD: Automated Insertion of Membrane Protein Structures into Explicit Lipid Membranes
- PMID: 26073602
- PMCID: PMC4509712
- DOI: 10.1016/j.str.2015.05.006
MemProtMD: Automated Insertion of Membrane Protein Structures into Explicit Lipid Membranes
Abstract
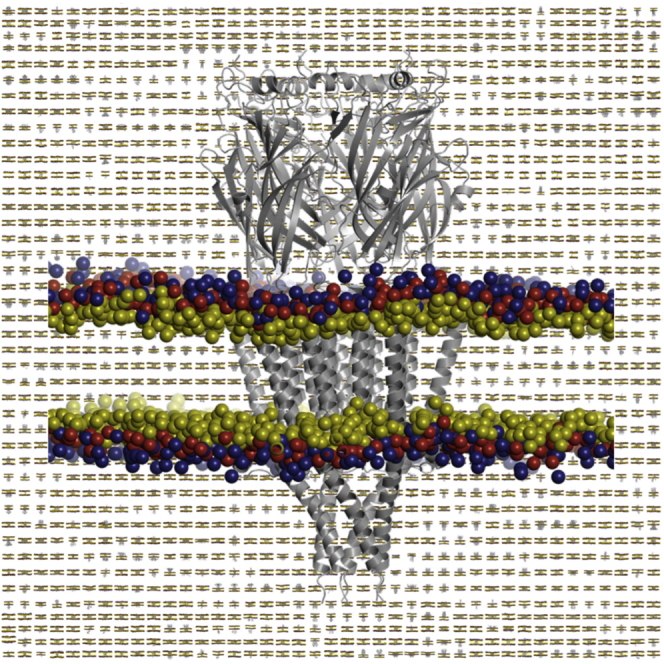
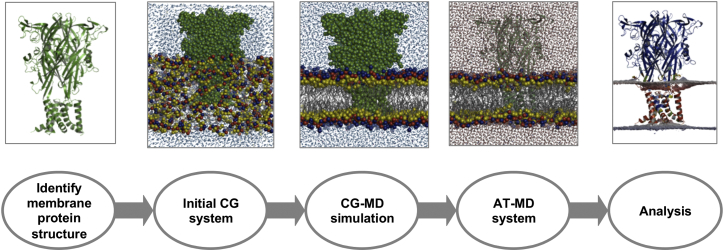
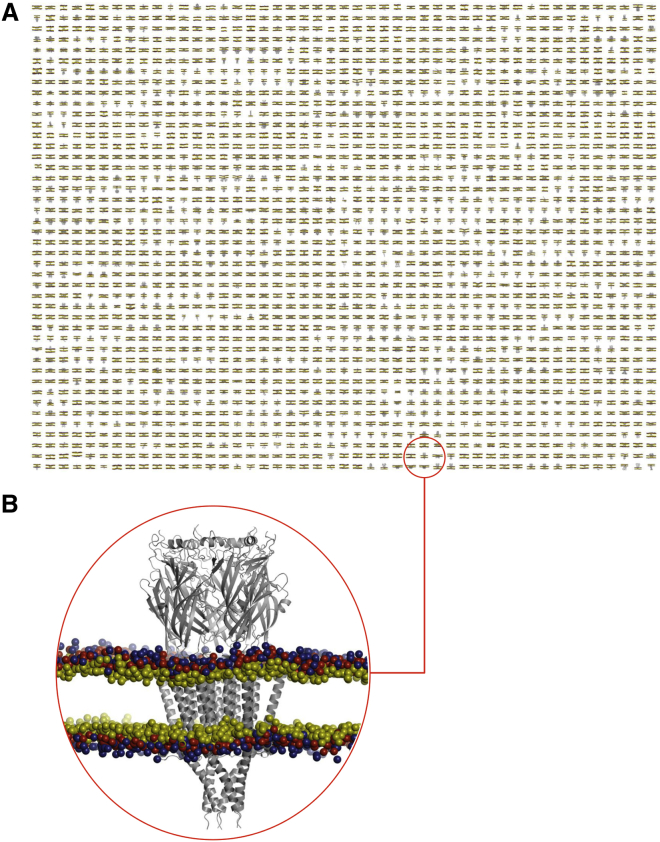
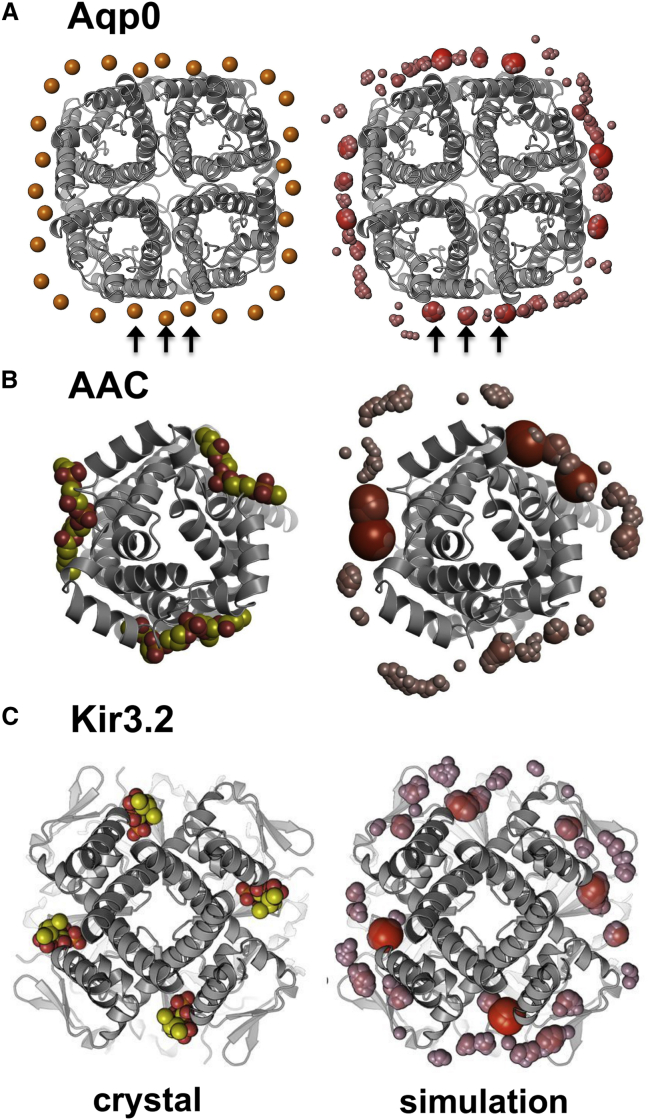
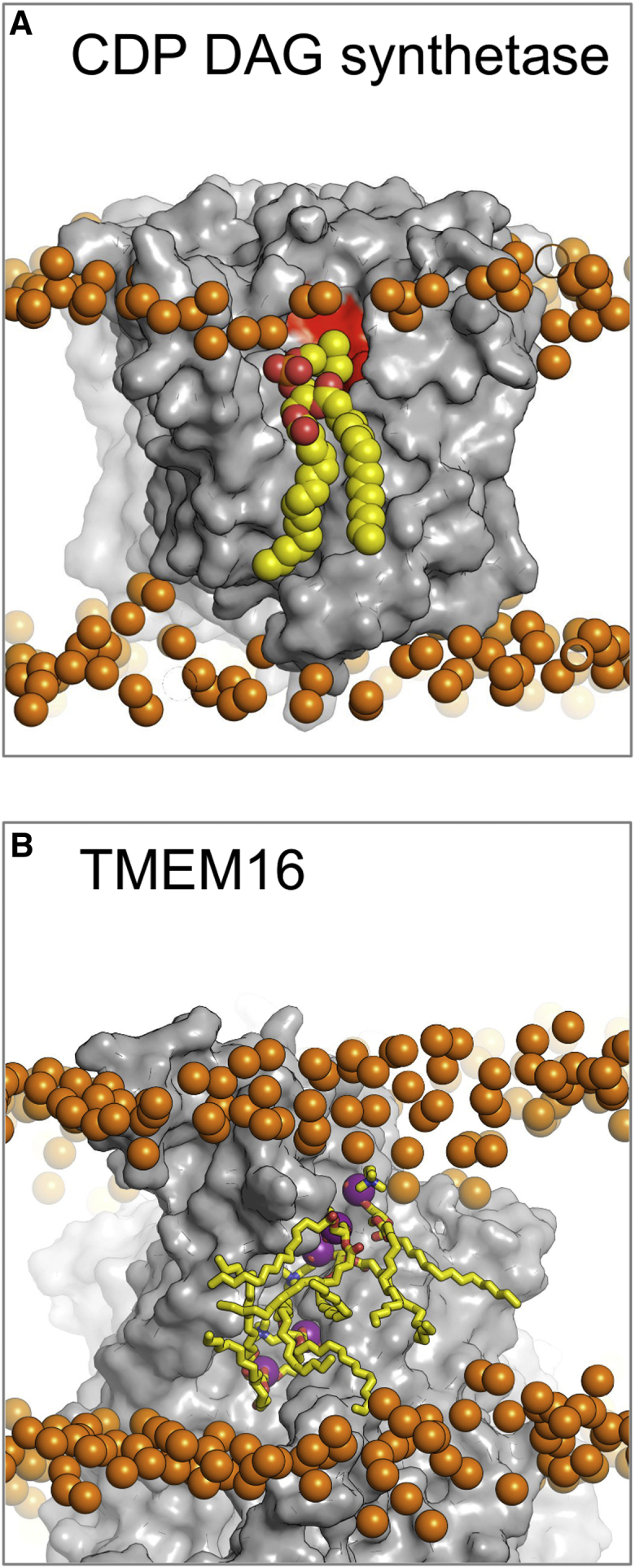
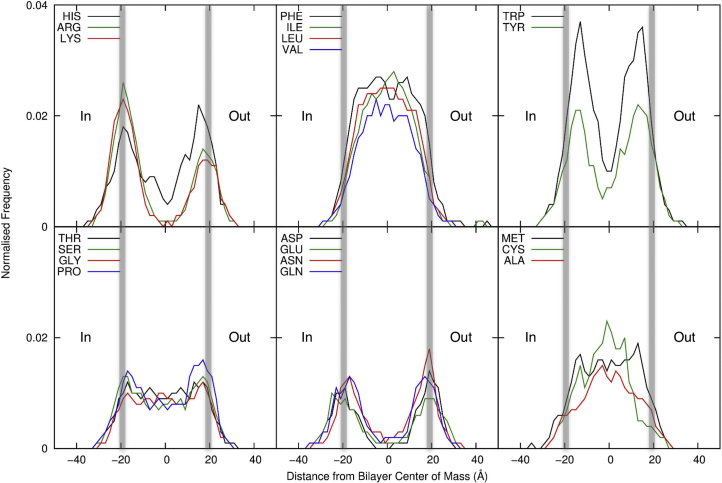
There has been exponential growth in the number of membrane protein structures determined. Nevertheless, these structures are usually resolved in the absence of their lipid environment. Coarse-grained molecular dynamics (CGMD) simulations enable insertion of membrane proteins into explicit models of lipid bilayers. We have automated the CGMD methodology, enabling membrane protein structures to be identified upon their release into the PDB and embedded into a membrane. The simulations are analyzed for protein-lipid interactions, identifying lipid binding sites, and revealing local bilayer deformations plus molecular access pathways within the membrane. The coarse-grained models of membrane protein/bilayer complexes are transformed to atomistic resolution for further analysis and simulation. Using this automated simulation pipeline, we have analyzed a number of recently determined membrane protein structures to predict their locations within a membrane, their lipid/protein interactions, and the functional implications of an enhanced understanding of the local membrane environment of each protein.
Copyright © 2015 The Authors. Published by Elsevier Ltd.. All rights reserved.
Figures



References
-
- Arnarez C., Mazat J.P., Elezgaray J., Marrink S.J., Periole X. Evidence for cardiolipin binding sites on the membrane-exposed surface of the cytochrome bc1. J. Am. Chem. Soc. 2013;135:3112–3120. - PubMed
-
- Badola P., Sanders C.R., 2nd Escherichia coli diacylglycerol kinase is an evolutionarily optimized membrane enzyme and catalyzes direct phosphoryl transfer. J. Biol. Chem. 1997;272:24176–24182. - PubMed
-
- Baeza-Delgado C., Marti-Renom M.A., Mingarro I. Structure-based statistical analysis of transmembrane helices. Eur. Biophys. J. 2013;42:199–207. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- BB/L002558/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/I019855/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BBS/B/16011/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BEP17032/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- 102890/Z/13/Z/WT_/Wellcome Trust/United Kingdom
- G0900399/MRC_/Medical Research Council/United Kingdom
- B19456/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/H000267/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- 106169/WT_/Wellcome Trust/United Kingdom
- BB/L002531/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- 084655/WT_/Wellcome Trust/United Kingdom
- 102890/WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
