

Soluble amyloid-β oligomers as synaptotoxins leading to cognitive impairment in Alzheimer's disease
- PMID: 26074767
- PMCID: PMC4443025
- DOI: 10.3389/fncel.2015.00191
Soluble amyloid-β oligomers as synaptotoxins leading to cognitive impairment in Alzheimer's disease
Abstract
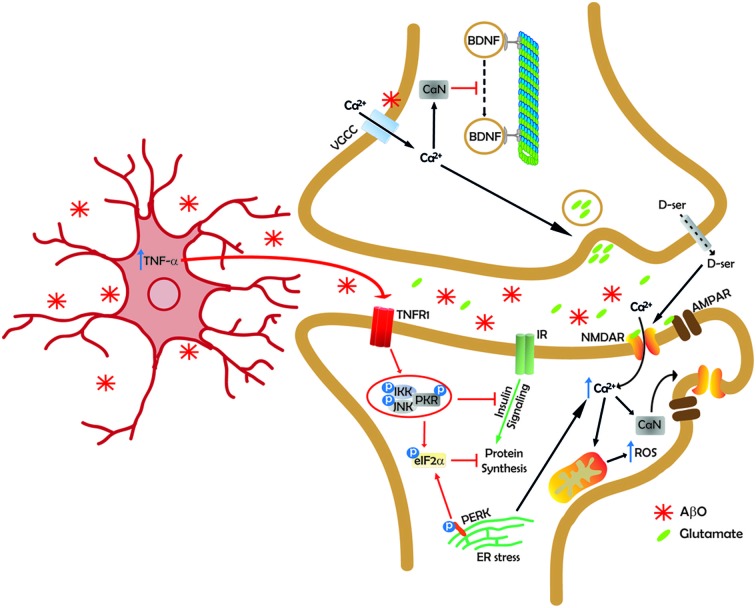
Alzheimer's disease (AD) is the most common form of dementia in the elderly, and affects millions of people worldwide. As the number of AD cases continues to increase in both developed and developing countries, finding therapies that effectively halt or reverse disease progression constitutes a major research and public health challenge. Since the identification of the amyloid-β peptide (Aβ) as the major component of the amyloid plaques that are characteristically found in AD brains, a major effort has aimed to determine whether and how Aβ leads to memory loss and cognitive impairment. A large body of evidence accumulated in the past 15 years supports a pivotal role of soluble Aβ oligomers (AβOs) in synapse failure and neuronal dysfunction in AD. Nonetheless, a number of basic questions, including the exact molecular composition of the synaptotoxic oligomers, the identity of the receptor(s) to which they bind, and the signaling pathways that ultimately lead to synapse failure, remain to be definitively answered. Here, we discuss recent advances that have illuminated our understanding of the chemical nature of the toxic species and the deleterious impact they have on synapses, and have culminated in the proposal of an Aβ oligomer hypothesis for Alzheimer's pathogenesis. We also highlight outstanding questions and challenges in AD research that should be addressed to allow translation of research findings into effective AD therapies.
Keywords: Alzheimer’s disease; amyloid-β oligomers; memory loss; neuronal dysfunction; synapse failure.
Figures
References
-
- Alzheimer A. (1907). Uber eine eigenartige Erkrankung der Hirnrinde. Allg. Z. Psychiatr. Phychish Gerichtl. Med. Berlin 64, 146–148.
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous