

A method to predict the impact of regulatory variants from DNA sequence
- PMID: 26075791
- PMCID: PMC4520745
- DOI: 10.1038/ng.3331
A method to predict the impact of regulatory variants from DNA sequence
Abstract
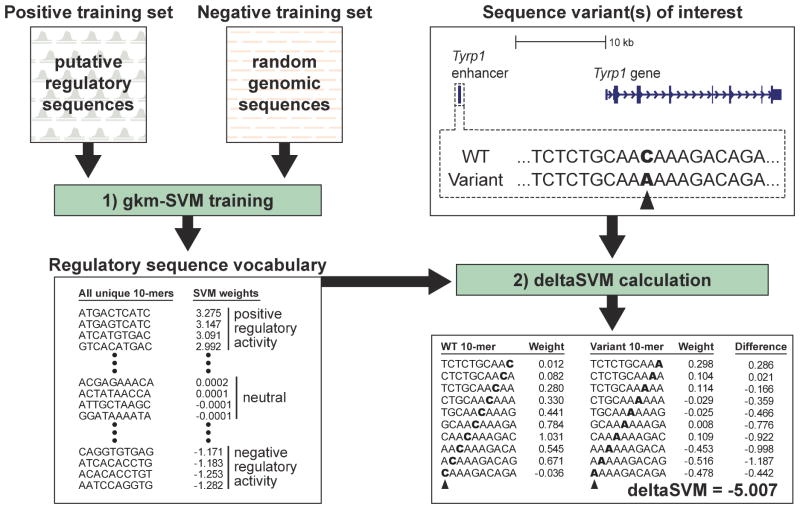
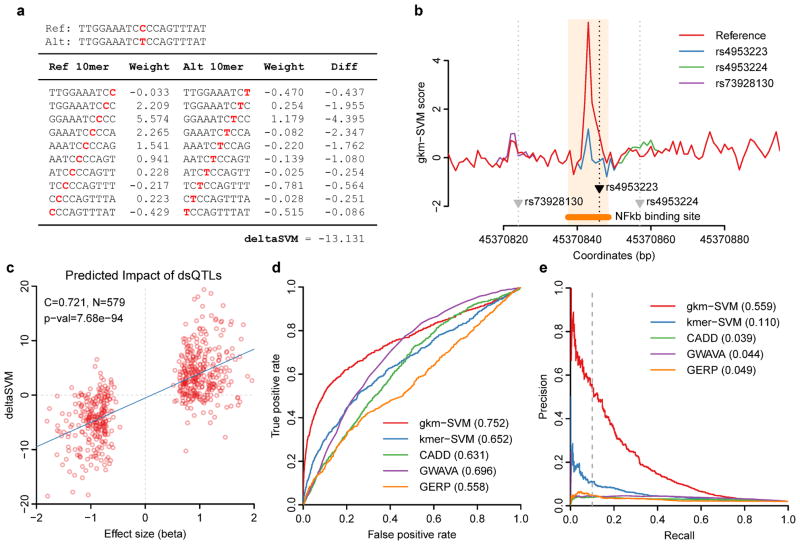
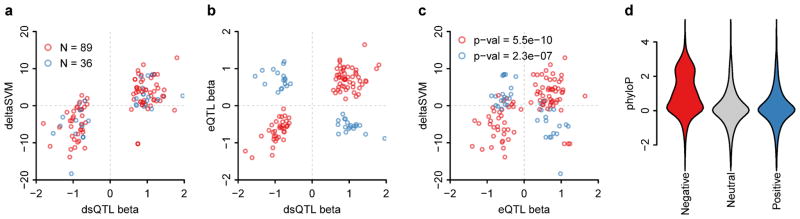
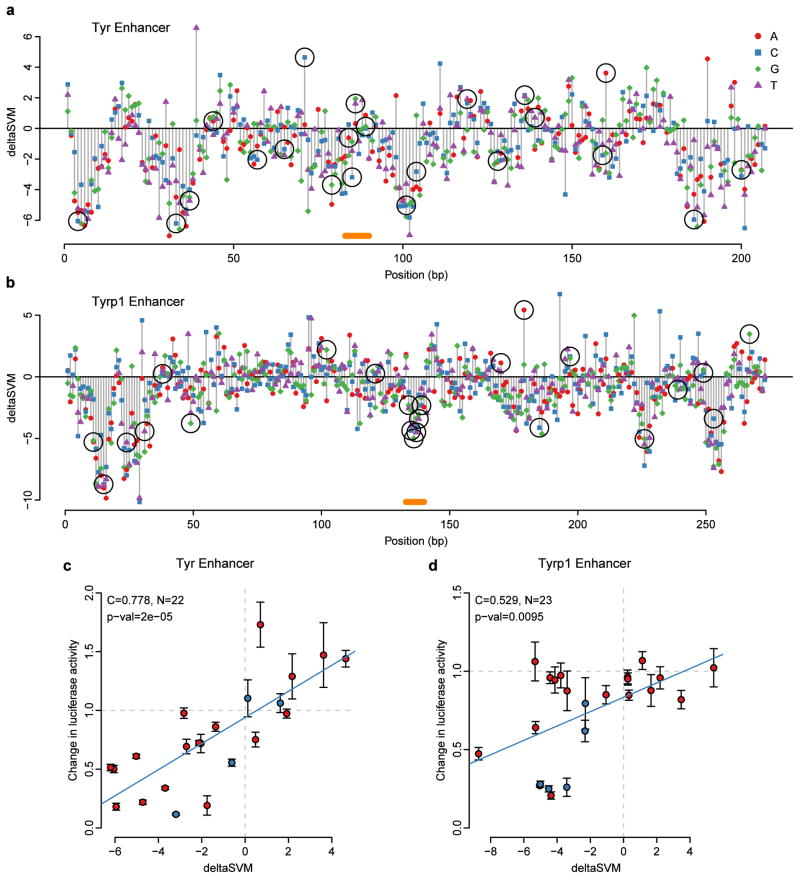
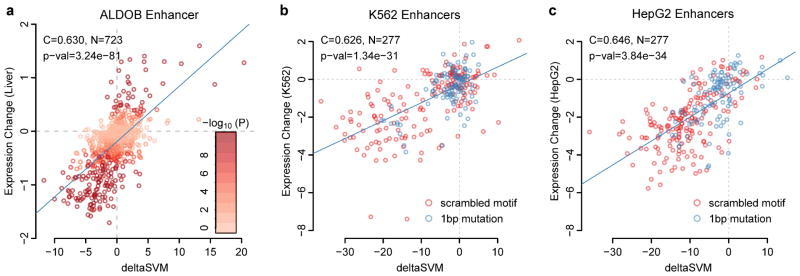
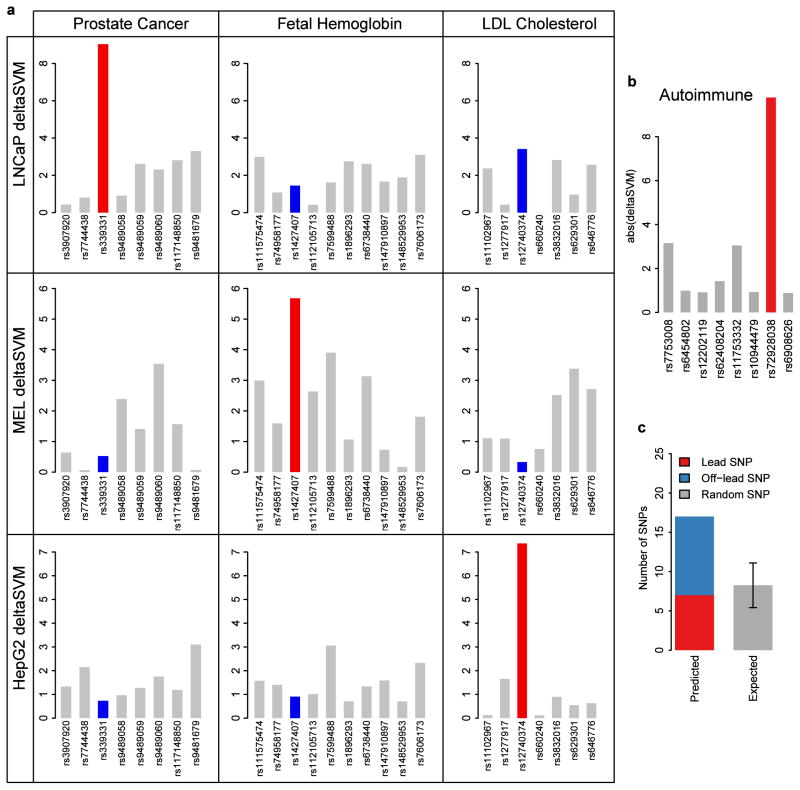
Most variants implicated in common human disease by genome-wide association studies (GWAS) lie in noncoding sequence intervals. Despite the suggestion that regulatory element disruption represents a common theme, identifying causal risk variants within implicated genomic regions remains a major challenge. Here we present a new sequence-based computational method to predict the effect of regulatory variation, using a classifier (gkm-SVM) that encodes cell type-specific regulatory sequence vocabularies. The induced change in the gkm-SVM score, deltaSVM, quantifies the effect of variants. We show that deltaSVM accurately predicts the impact of SNPs on DNase I sensitivity in their native genomic contexts and accurately predicts the results of dense mutagenesis of several enhancers in reporter assays. Previously validated GWAS SNPs yield large deltaSVM scores, and we predict new risk-conferring SNPs for several autoimmune diseases. Thus, deltaSVM provides a powerful computational approach to systematically identify functional regulatory variants.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
Comment in
-
Running spell-check to identify regulatory variants.Nat Genet. 2015 Aug;47(8):853-5. doi: 10.1038/ng.3364. Nat Genet. 2015. PMID: 26220134
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
