

A mass-tolerant database search identifies a large proportion of unassigned spectra in shotgun proteomics as modified peptides
- PMID: 26076430
- PMCID: PMC4515955
- DOI: 10.1038/nbt.3267
A mass-tolerant database search identifies a large proportion of unassigned spectra in shotgun proteomics as modified peptides
Erratum in
- Nat Biotechnol. 2015 Aug;33(8):882
-
Erratum: A mass-tolerant database search identifies a large proportion of unassigned spectra in shotgun proteomics as modified peptides.Nat Biotechnol. 2015 Aug;33(8):882. doi: 10.1038/nbt0815-882d. Nat Biotechnol. 2015. PMID: 26252148 No abstract available.
Abstract
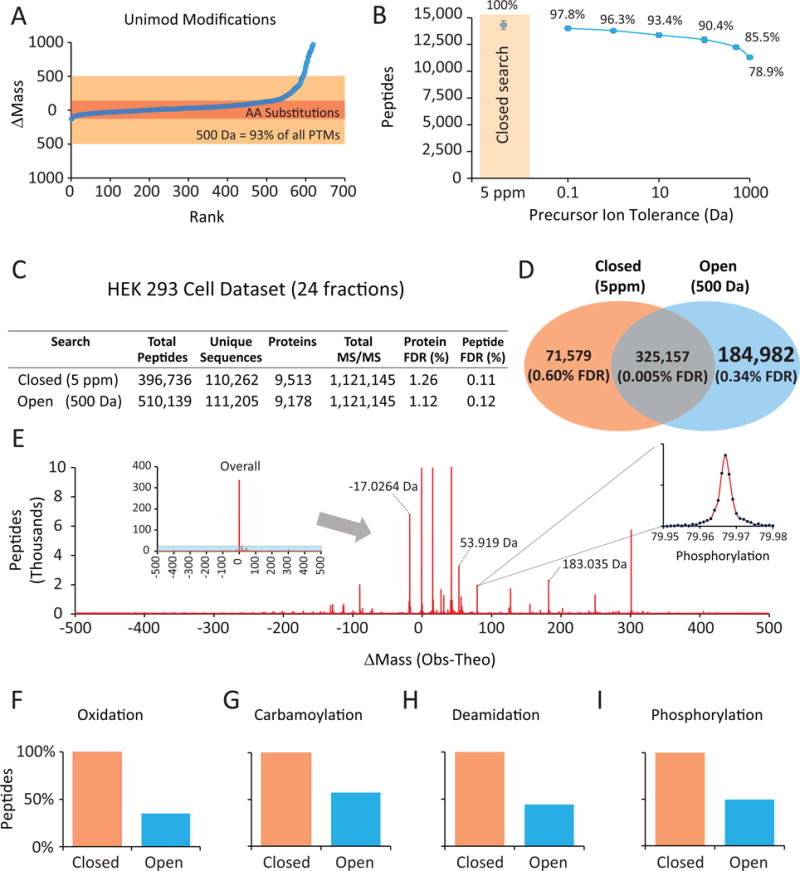
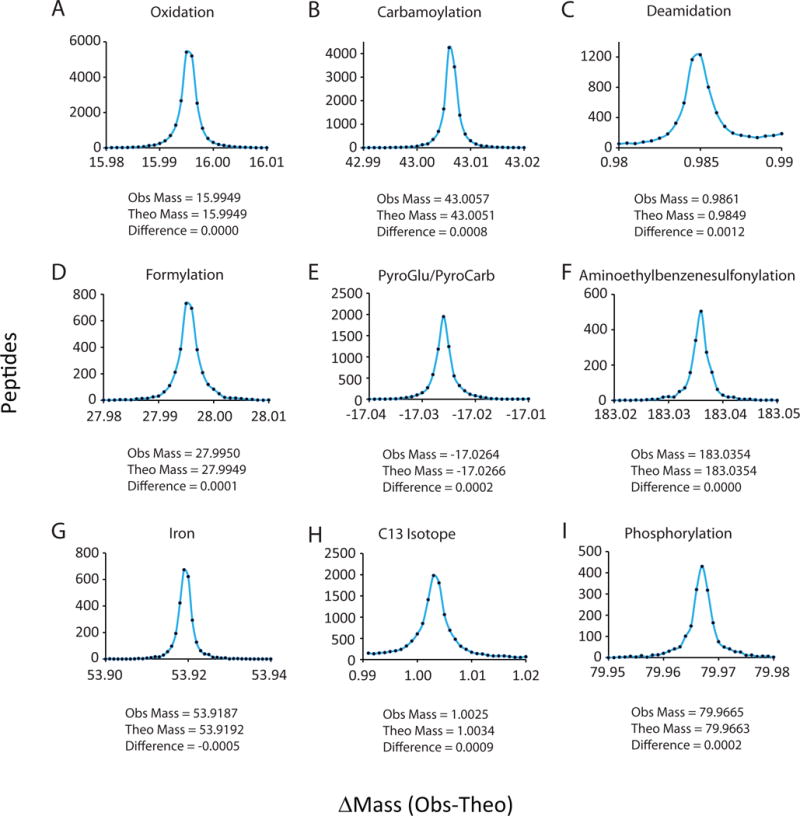
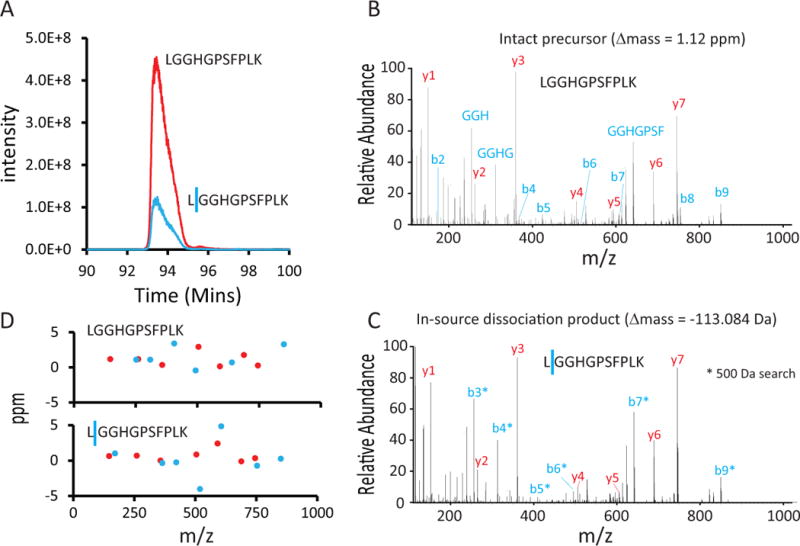
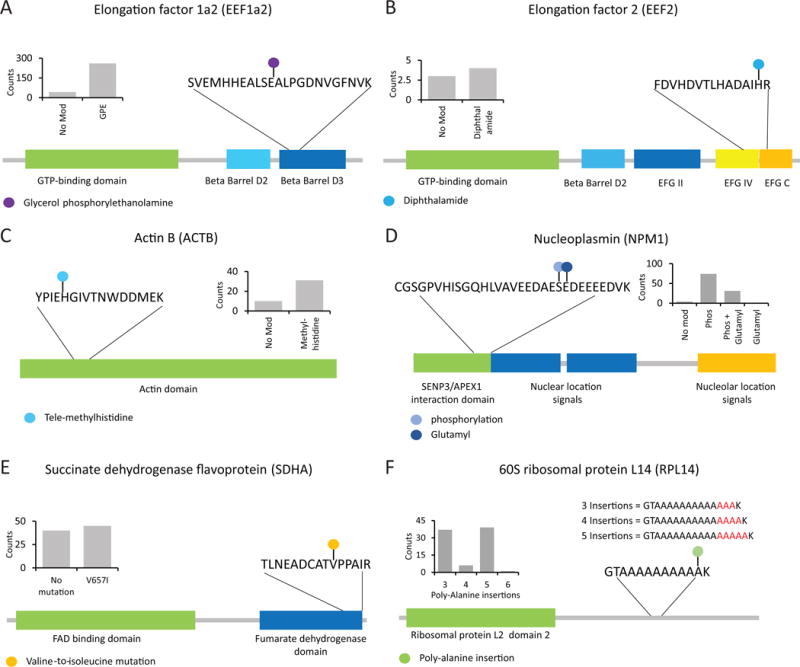
Fewer than half of all tandem mass spectrometry (MS/MS) spectra acquired in shotgun proteomics experiments are typically matched to a peptide with high confidence. Here we determine the identity of unassigned peptides using an ultra-tolerant Sequest database search that allows peptide matching even with modifications of unknown masses up to ± 500 Da. In a proteome-wide data set on HEK293 cells (9,513 proteins and 396,736 peptides), this approach matched an additional 184,000 modified peptides, which were linked to biological and chemical modifications representing 523 distinct mass bins, including phosphorylation, glycosylation and methylation. We localized all unknown modification masses to specific regions within a peptide. Known modifications were assigned to the correct amino acids with frequencies >90%. We conclude that at least one-third of unassigned spectra arise from peptides with substoichiometric modifications.
Figures
Comment in
-
Illuminating the dark matter of shotgun proteomics.Nat Biotechnol. 2015 Jul;33(7):717-8. doi: 10.1038/nbt.3287. Nat Biotechnol. 2015. PMID: 26154010 No abstract available.
References
-
- Washburn MP, Wolters D, Yates JR., 3rd Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol. 2001;19:242–247. - PubMed
-
- Wolters DA, Washburn MP, Yates JR., 3rd An automated multidimensional protein identification technology for shotgun proteomics. Anal Chem. 2001;73:5683–5690. - PubMed
-
- Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom. 1994;5:976–89. - PubMed
-
- Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
