

Parkinson-causing α-synuclein missense mutations shift native tetramers to monomers as a mechanism for disease initiation
- PMID: 26076669
- PMCID: PMC4490410
- DOI: 10.1038/ncomms8314
Parkinson-causing α-synuclein missense mutations shift native tetramers to monomers as a mechanism for disease initiation
Erratum in
-
Corrigendum: Parkinson-causing α-synuclein missense mutations shift native tetramers to monomers as a mechanism for disease initiation.Nat Commun. 2015 Jul 30;6:8008. doi: 10.1038/ncomms9008. Nat Commun. 2015. PMID: 26224447 Free PMC article. No abstract available.
Abstract
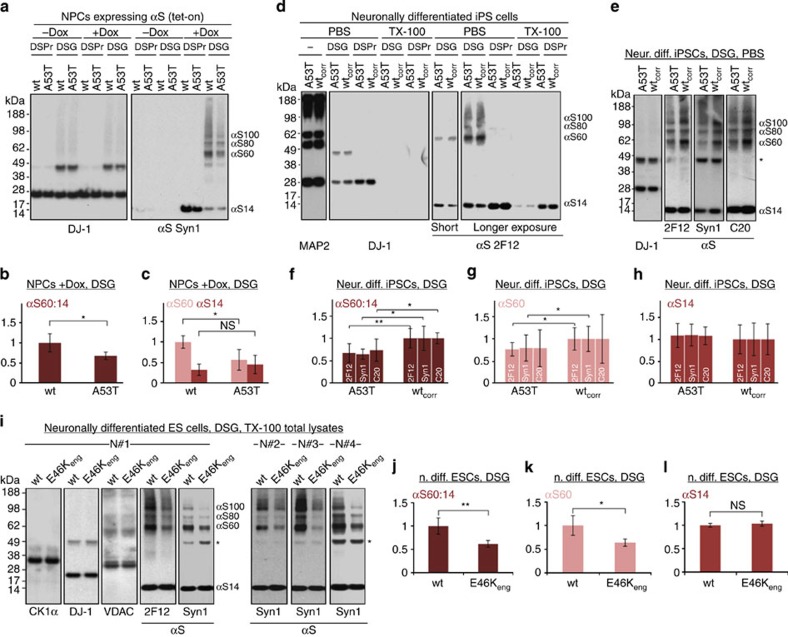
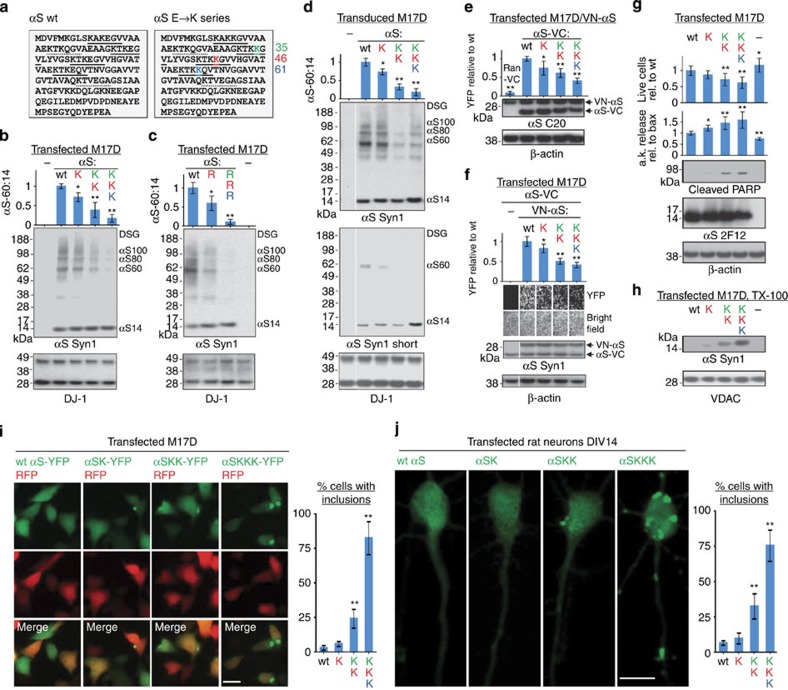
β-Sheet-rich α-synuclein (αS) aggregates characterize Parkinson's disease (PD). αS was long believed to be a natively unfolded monomer, but recent work suggests it also occurs in α-helix-rich tetramers. Crosslinking traps principally tetrameric αS in intact normal neurons, but not after cell lysis, suggesting a dynamic equilibrium. Here we show that freshly biopsied normal human brain contains abundant αS tetramers. The PD-causing mutation A53T decreases tetramers in mouse brain. Neurons derived from an A53T patient have decreased tetramers. Neurons expressing E46K do also, and adding 1-2 E46K-like mutations into the canonical αS repeat motifs (KTKEGV) further reduces tetramers, decreases αS solubility and induces neurotoxicity and round inclusions. The other three fPD missense mutations likewise decrease tetramer:monomer ratios. The destabilization of physiological tetramers by PD-causing missense mutations and the neurotoxicity and inclusions induced by markedly decreasing tetramers suggest that decreased α-helical tetramers and increased unfolded monomers initiate pathogenesis. Tetramer-stabilizing compounds should prevent this.
Conflict of interest statement
D.S. is a director and consultant to Prothena Biosciences. The remaining authors declare no competing financial interests.
Figures
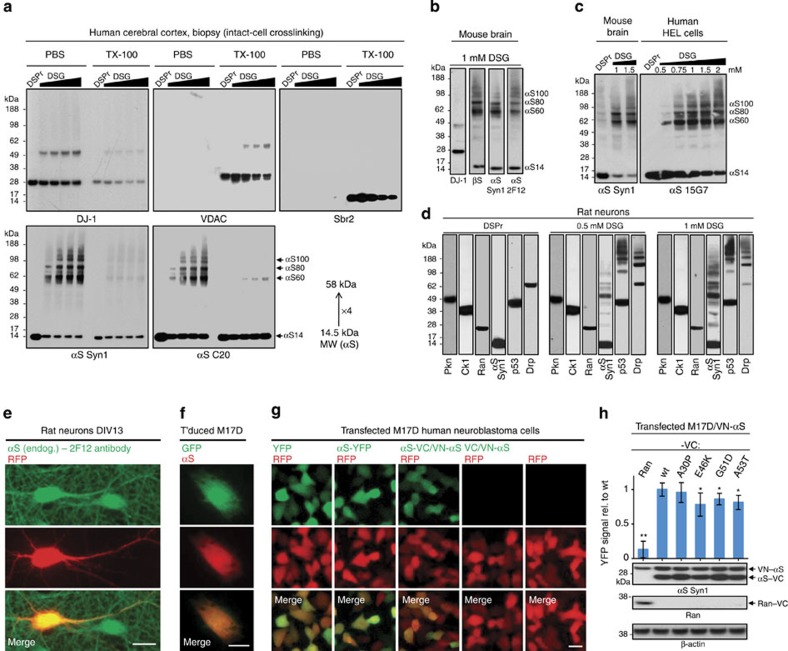
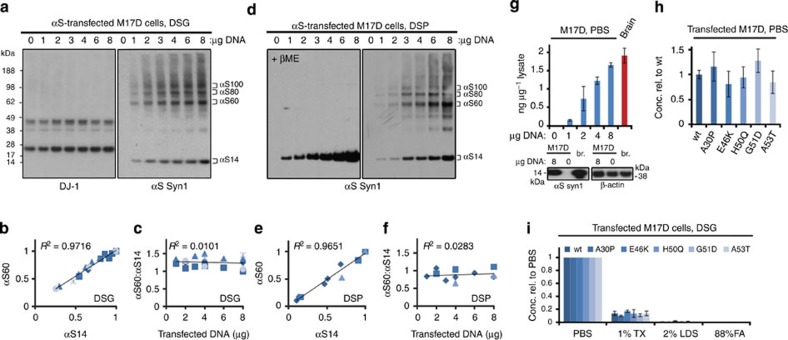
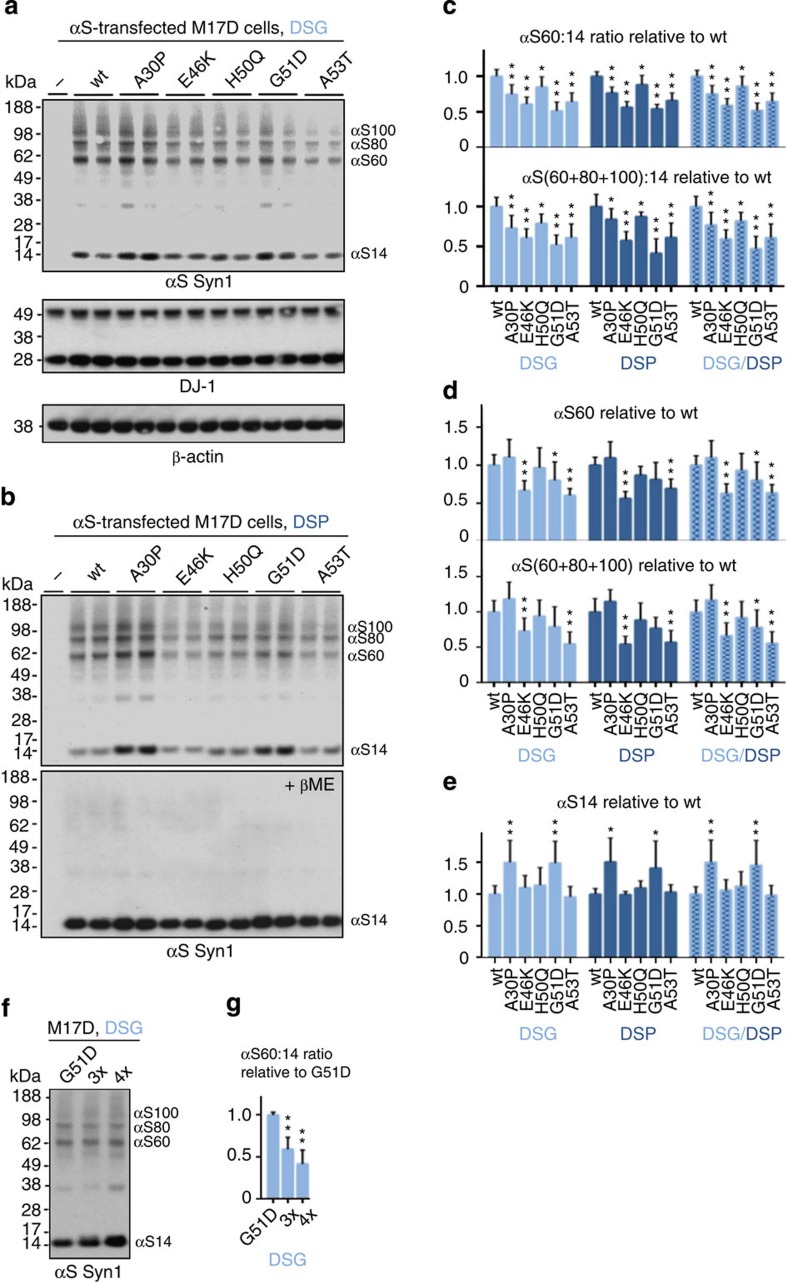
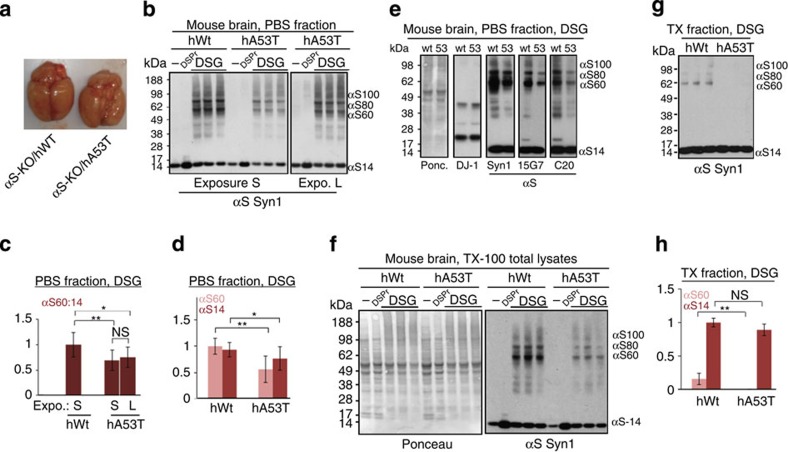
Comment in
-
Parkinson disease: Disease-linked morphometric variants of α-synuclein.Nat Rev Neurol. 2015 Aug;11(8):428. doi: 10.1038/nrneurol.2015.118. Epub 2015 Jul 7. Nat Rev Neurol. 2015. PMID: 26149979 No abstract available.
References
-
- Spillantini M. G. et al.. Alpha-synuclein in Lewy bodies. Nature 388, 839–840 (1997). - PubMed
-
- Polymeropoulos M. H. et al.. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 276, 2045–2047 (1997). - PubMed
-
- Krüger R. et al.. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat. Genet. 18, 106–108 (1998). - PubMed
-
- Zarranz J. J. et al.. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 55, 164–173 (2004). - PubMed
-
- Singleton A. B. et al.. alpha-Synuclein locus triplication causes Parkinson's disease. Science 302, 841 (2003). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
