

Statistically based splicing detection reveals neural enrichment and tissue-specific induction of circular RNA during human fetal development
- PMID: 26076956
- PMCID: PMC4506483
- DOI: 10.1186/s13059-015-0690-5
Statistically based splicing detection reveals neural enrichment and tissue-specific induction of circular RNA during human fetal development
Erratum in
-
Erratum to: Statistically based splicing detection reveals neural enrichment and tissue-specific induction of circular RNA during human fetal development.Genome Biol. 2016 Dec 19;17(1):263. doi: 10.1186/s13059-016-1123-9. Genome Biol. 2016. PMID: 27993159 Free PMC article. No abstract available.
Abstract
Background: The pervasive expression of circular RNA is a recently discovered feature of gene expression in highly diverged eukaryotes, but the functions of most circular RNAs are still unknown. Computational methods to discover and quantify circular RNA are essential. Moreover, discovering biological contexts where circular RNAs are regulated will shed light on potential functional roles they may play.
Results: We present a new algorithm that increases the sensitivity and specificity of circular RNA detection by discovering and quantifying circular and linear RNA splicing events at both annotated and un-annotated exon boundaries, including intergenic regions of the genome, with high statistical confidence. Unlike approaches that rely on read count and exon homology to determine confidence in prediction of circular RNA expression, our algorithm uses a statistical approach. Using our algorithm, we unveiled striking induction of general and tissue-specific circular RNAs, including in the heart and lung, during human fetal development. We discover regions of the human fetal brain, such as the frontal cortex, with marked enrichment for genes where circular RNA isoforms are dominant.
Conclusions: The vast majority of circular RNA production occurs at major spliceosome splice sites; however, we find the first examples of developmentally induced circular RNAs processed by the minor spliceosome, and an enriched propensity of minor spliceosome donors to splice into circular RNA at un-annotated, rather than annotated, exons. Together, these results suggest a potentially significant role for circular RNA in human development.
Figures
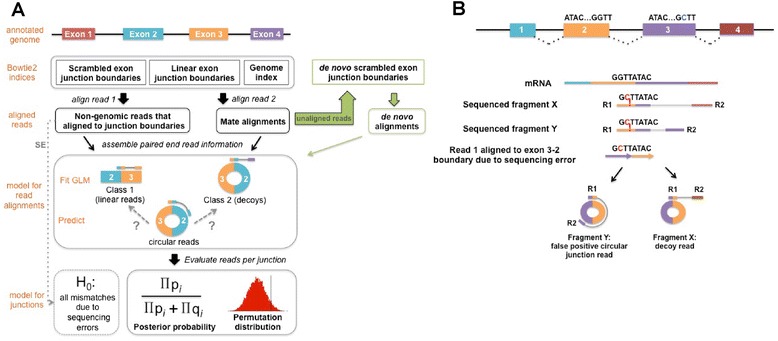
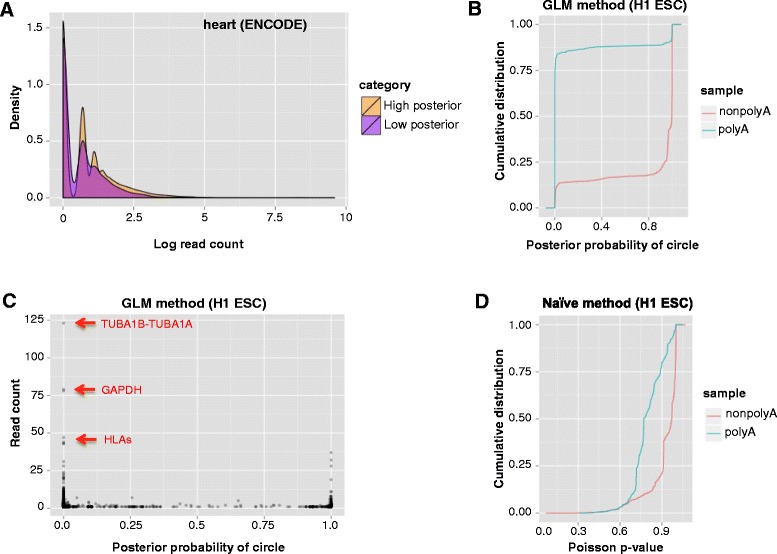
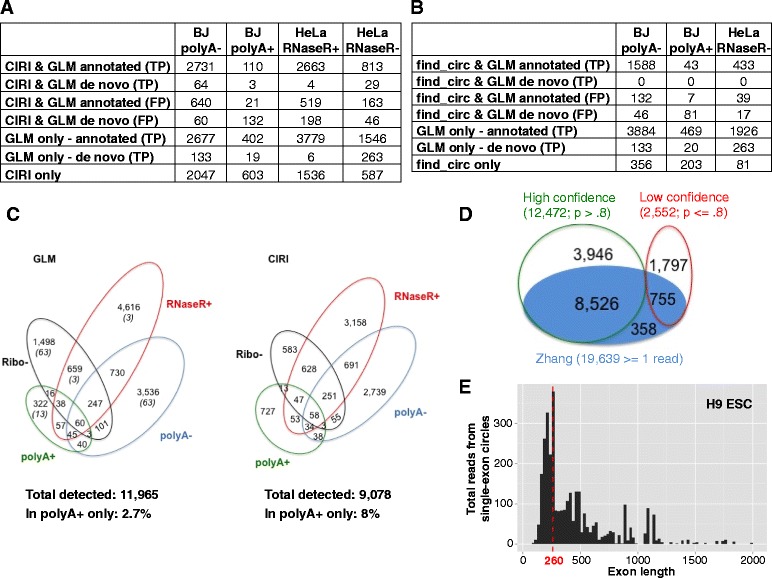
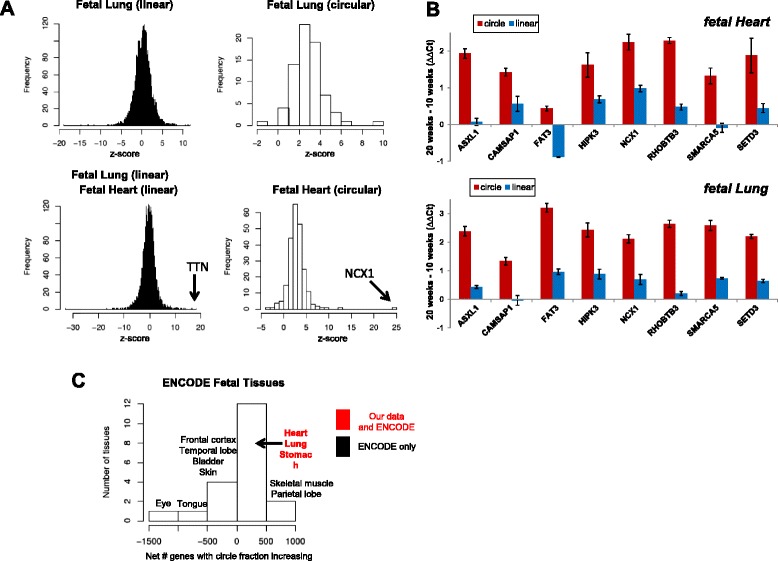
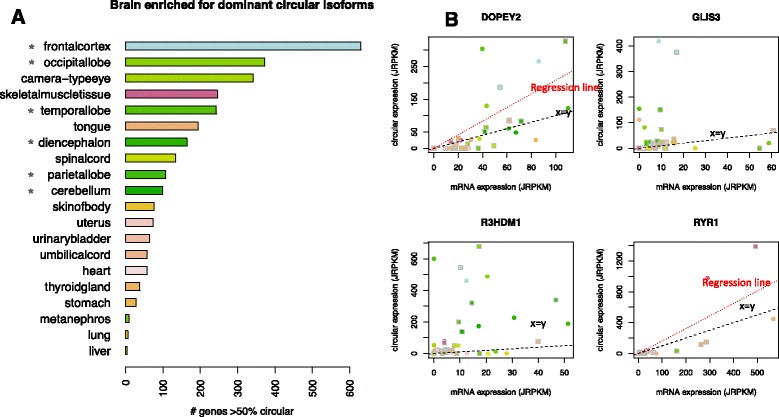
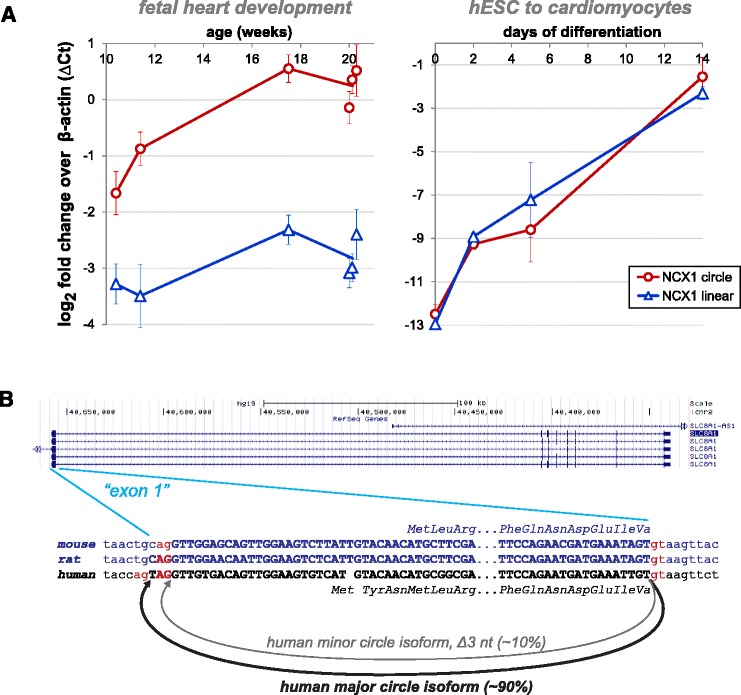
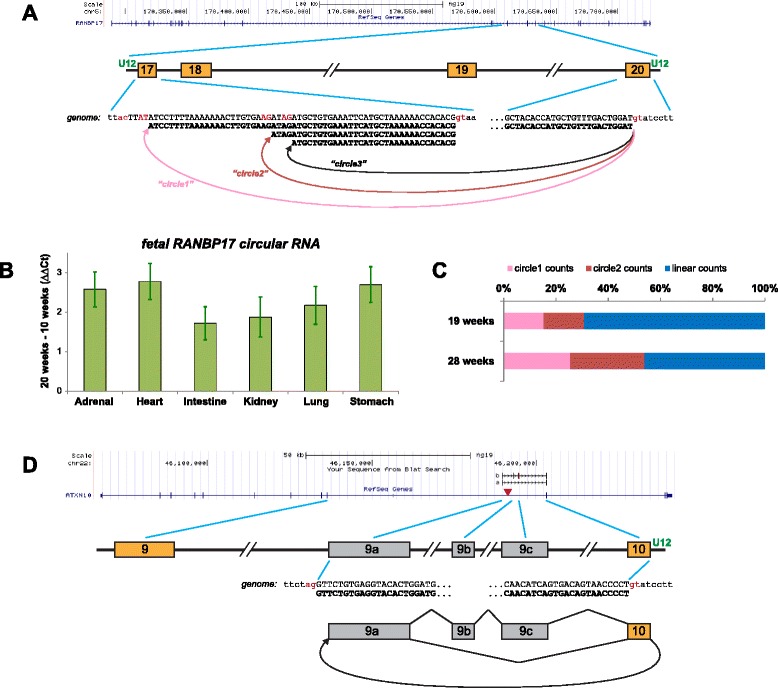
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
