

Anchor-based classification and type-C inhibitors for tyrosine kinases
- PMID: 26077136
- PMCID: PMC4468516
- DOI: 10.1038/srep10938
Anchor-based classification and type-C inhibitors for tyrosine kinases
Abstract
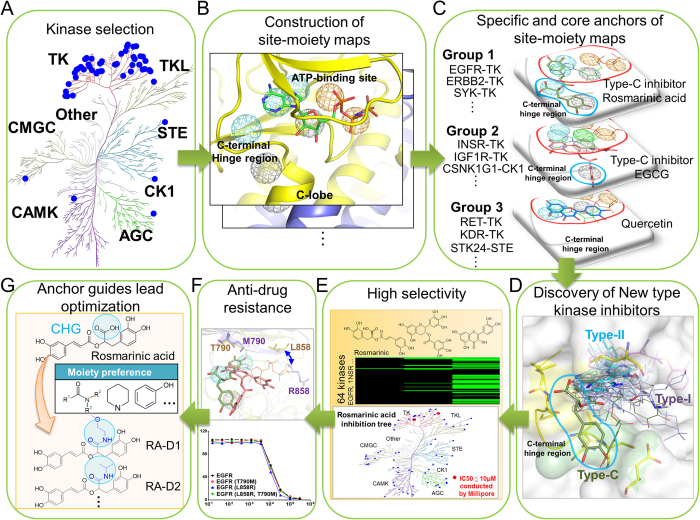
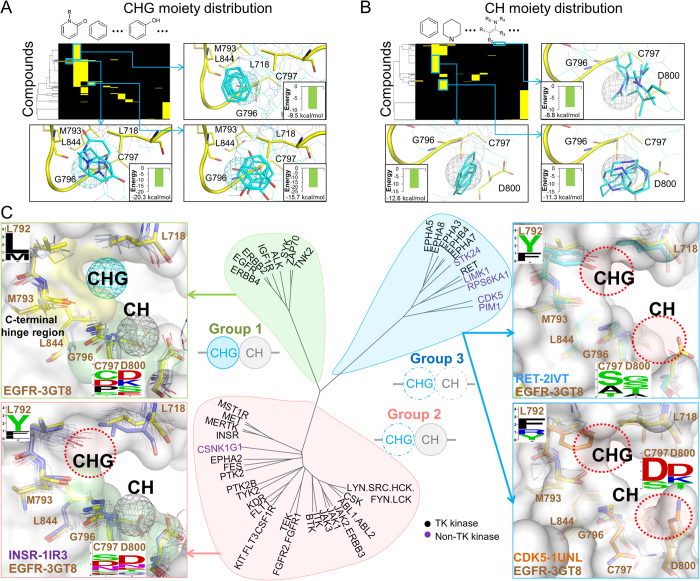
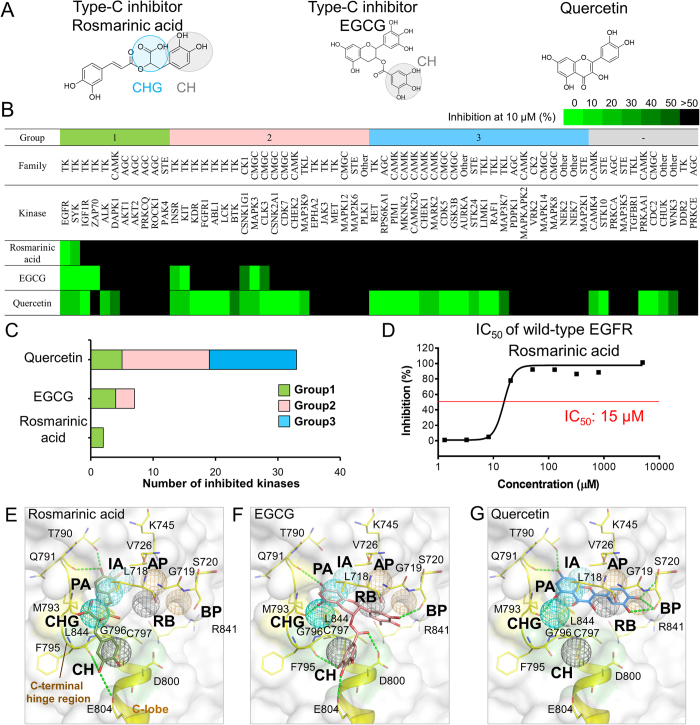
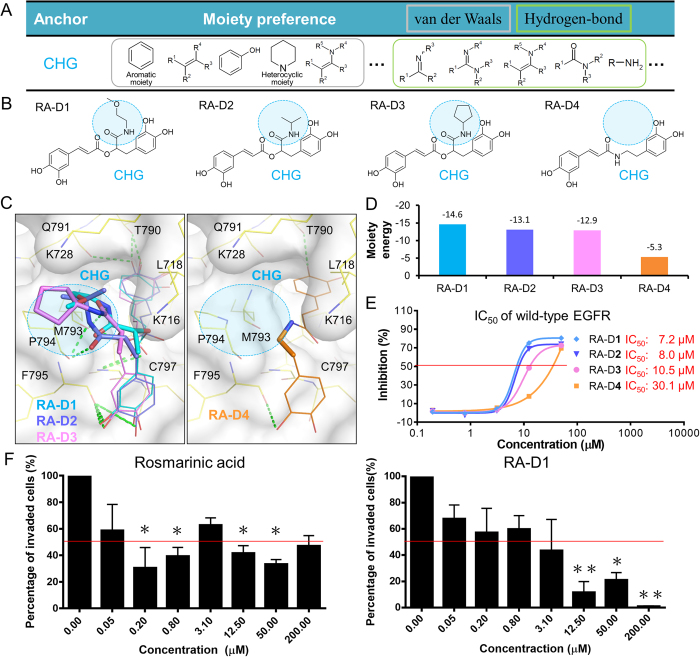
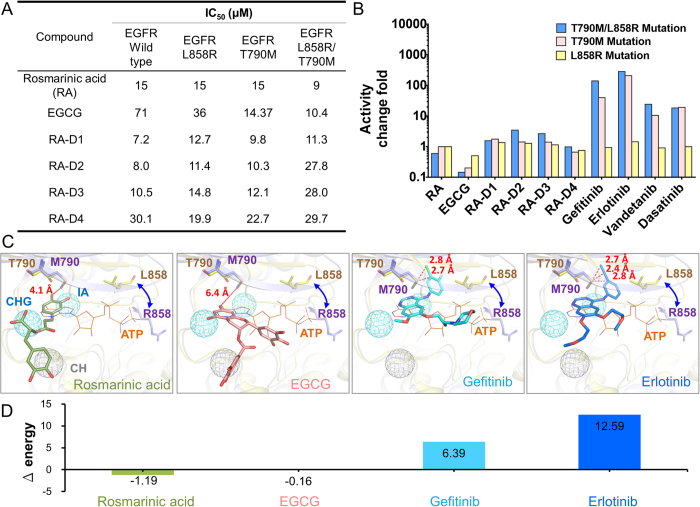
Tyrosine kinases regulate various biological processes and are drug targets for cancers. At present, the design of selective and anti-resistant inhibitors of kinases is an emergent task. Here, we inferred specific site-moiety maps containing two specific anchors to uncover a new binding pocket in the C-terminal hinge region by docking 4,680 kinase inhibitors into 51 protein kinases, and this finding provides an opportunity for the development of kinase inhibitors with high selectivity and anti-drug resistance. We present an anchor-based classification for tyrosine kinases and discover two type-C inhibitors, namely rosmarinic acid (RA) and EGCG, which occupy two and one specific anchors, respectively, by screening 118,759 natural compounds. Our profiling reveals that RA and EGCG selectively inhibit 3% (EGFR and SYK) and 14% of 64 kinases, respectively. According to the guide of our anchor model, we synthesized three RA derivatives with better potency. These type-C inhibitors are able to maintain activities for drug-resistant EGFR and decrease the invasion ability of breast cancer cells. Our results show that the type-C inhibitors occupying a new pocket are promising for cancer treatments due to their kinase selectivity and anti-drug resistance.
Figures
References
-
- Endicott J. A., Noble M. E. M. & Johnson L. N. The Structural Basis for Control of Eukaryotic Protein Kinases. Annu. Rev. Biochem. 81, 587–613 (2012). - PubMed
-
- Levitzki A. Tyrosine Kinase Inhibitors: Views of Selectivity, Sensitivity, and Clinical Performance. Annu. Rev. Pharmacol. Toxicol. 53, 161–185 (2013). - PubMed
-
- Krause D. S. & Van Etten R. A. Tyrosine kinases as targets for cancer therapy. N. Engl. J. Med. 353, 172–187 (2005). - PubMed
-
- Gocek E., Moulas A. N. & Studzinski G. P. Non-receptor protein tyrosine kinases signaling pathways in normal and cancer cells. Crit. Rev. Clin. Lab. Sci. 51, 125–137 (2014). - PubMed
-
- Liao J. J. L. Molecular recognition of protein kinase binding pockets for design of potent and selective kinase inhibitors. J. Med. Chem. 50, 409–424 (2007). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
