

Next-generation libraries for robust RNA interference-based genome-wide screens
- PMID: 26080438
- PMCID: PMC4491794
- DOI: 10.1073/pnas.1508821112
Next-generation libraries for robust RNA interference-based genome-wide screens
Abstract
Genetic screening based on loss-of-function phenotypes is a powerful discovery tool in biology. Although the recent development of clustered regularly interspaced short palindromic repeats (CRISPR)-based screening approaches in mammalian cell culture has enormous potential, RNA interference (RNAi)-based screening remains the method of choice in several biological contexts. We previously demonstrated that ultracomplex pooled short-hairpin RNA (shRNA) libraries can largely overcome the problem of RNAi off-target effects in genome-wide screens. Here, we systematically optimize several aspects of our shRNA library, including the promoter and microRNA context for shRNA expression, selection of guide strands, and features relevant for postscreen sample preparation for deep sequencing. We present next-generation high-complexity libraries targeting human and mouse protein-coding genes, which we grouped into 12 sublibraries based on biological function. A pilot screen suggests that our next-generation RNAi library performs comparably to current CRISPR interference (CRISPRi)-based approaches and can yield complementary results with high sensitivity and high specificity.
Keywords: functional genomics; genetic screen; microRNA; pooled screen; shRNA.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
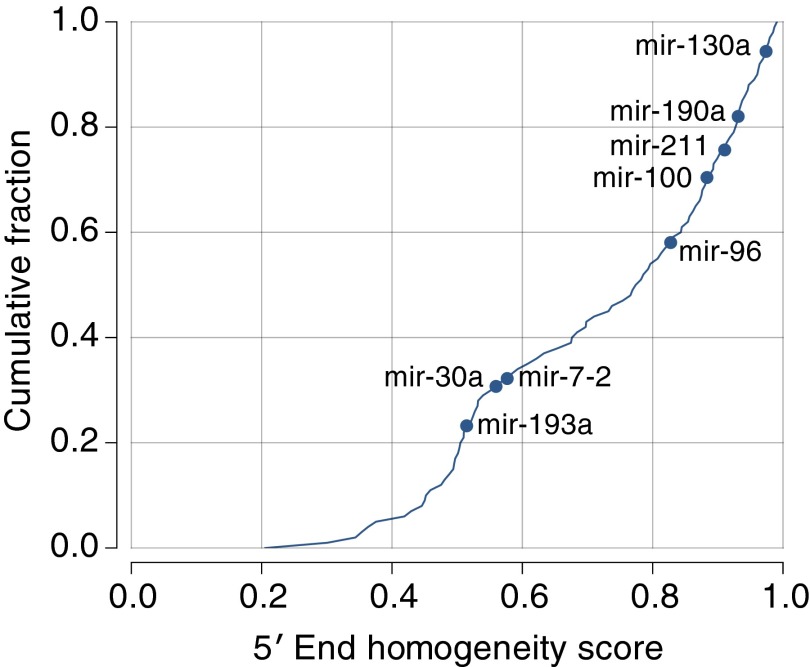
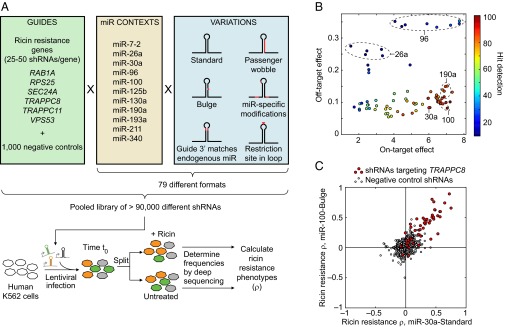
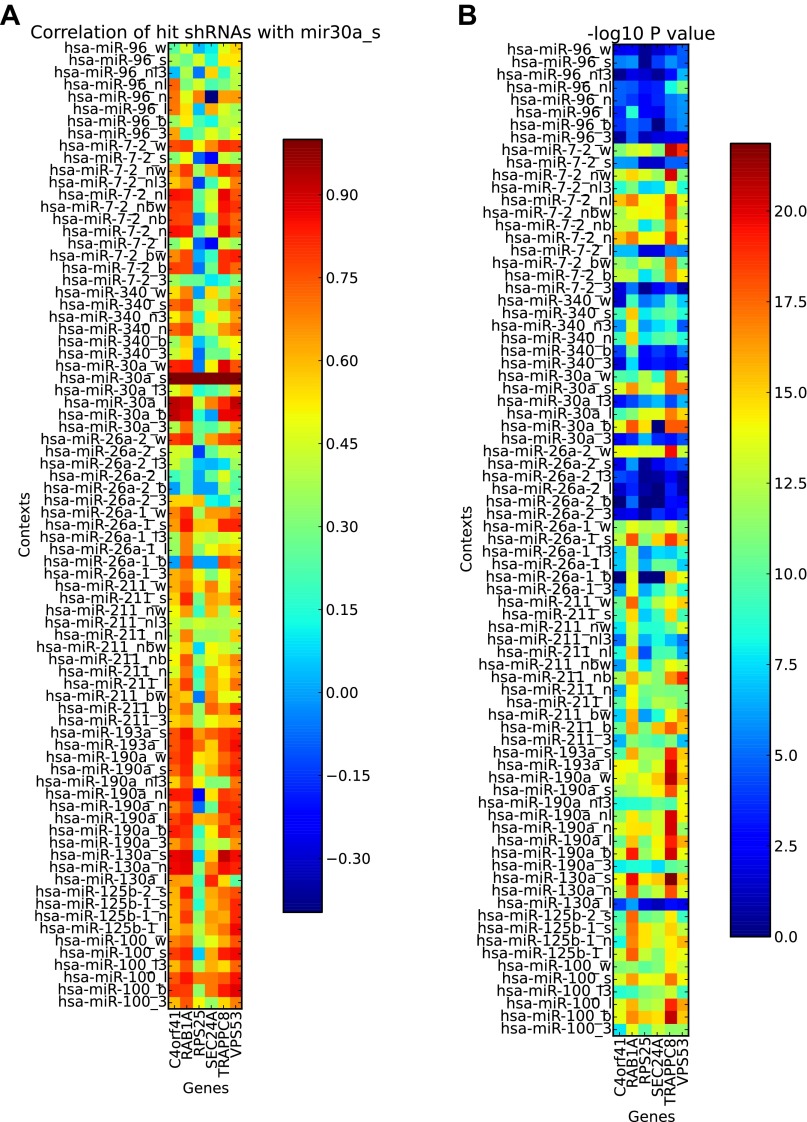
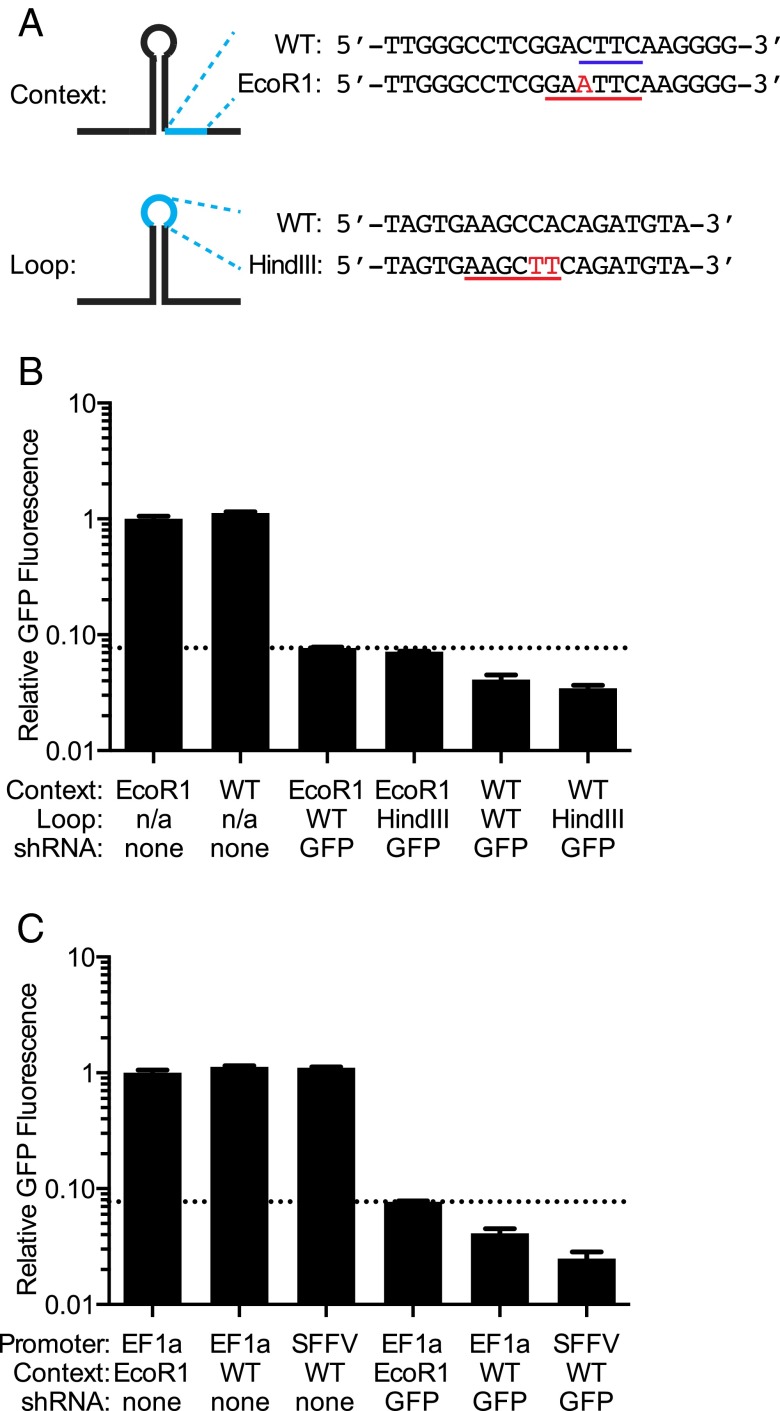
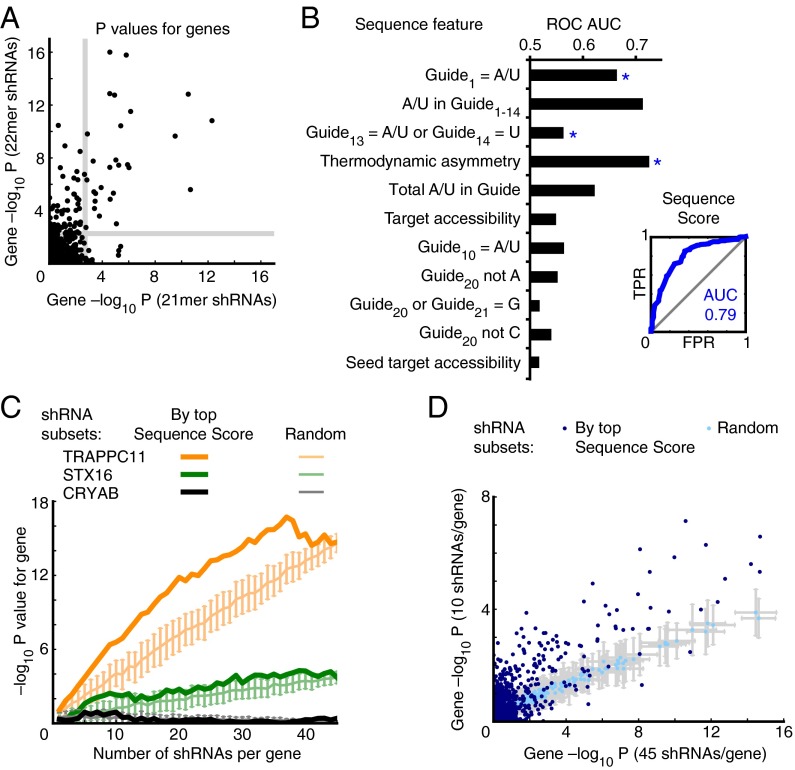
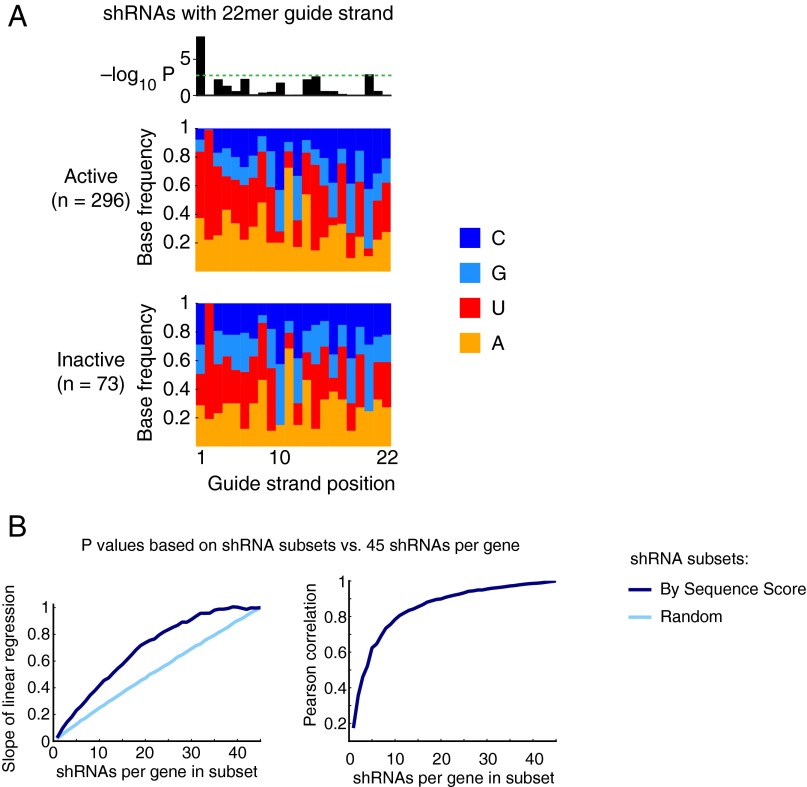
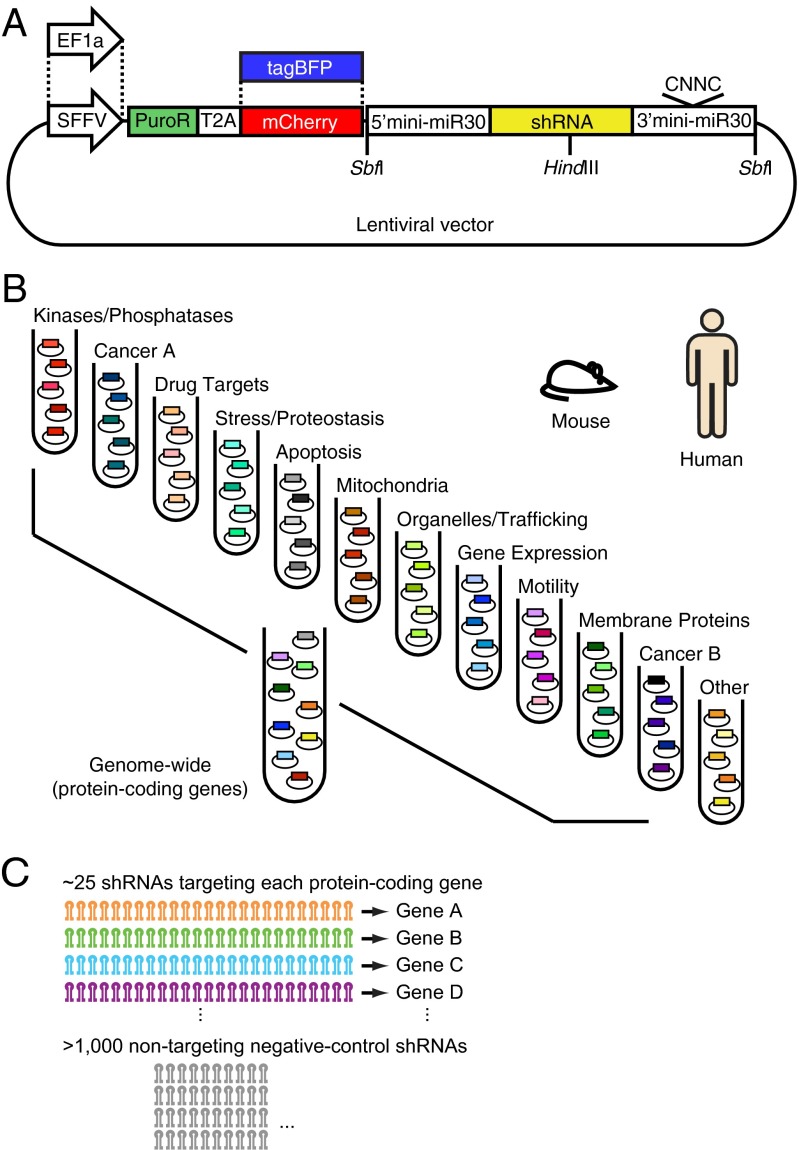
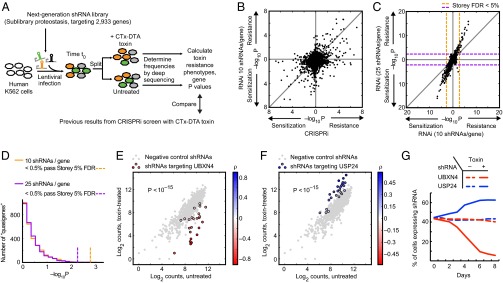
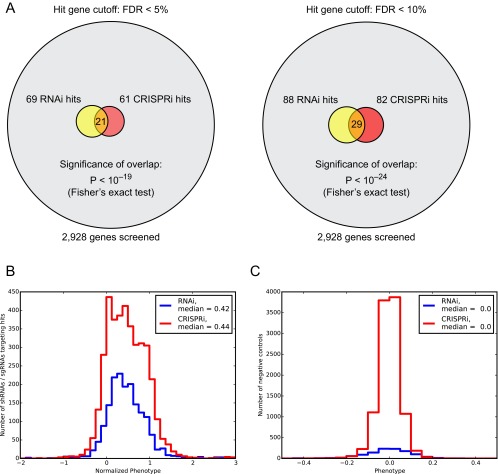
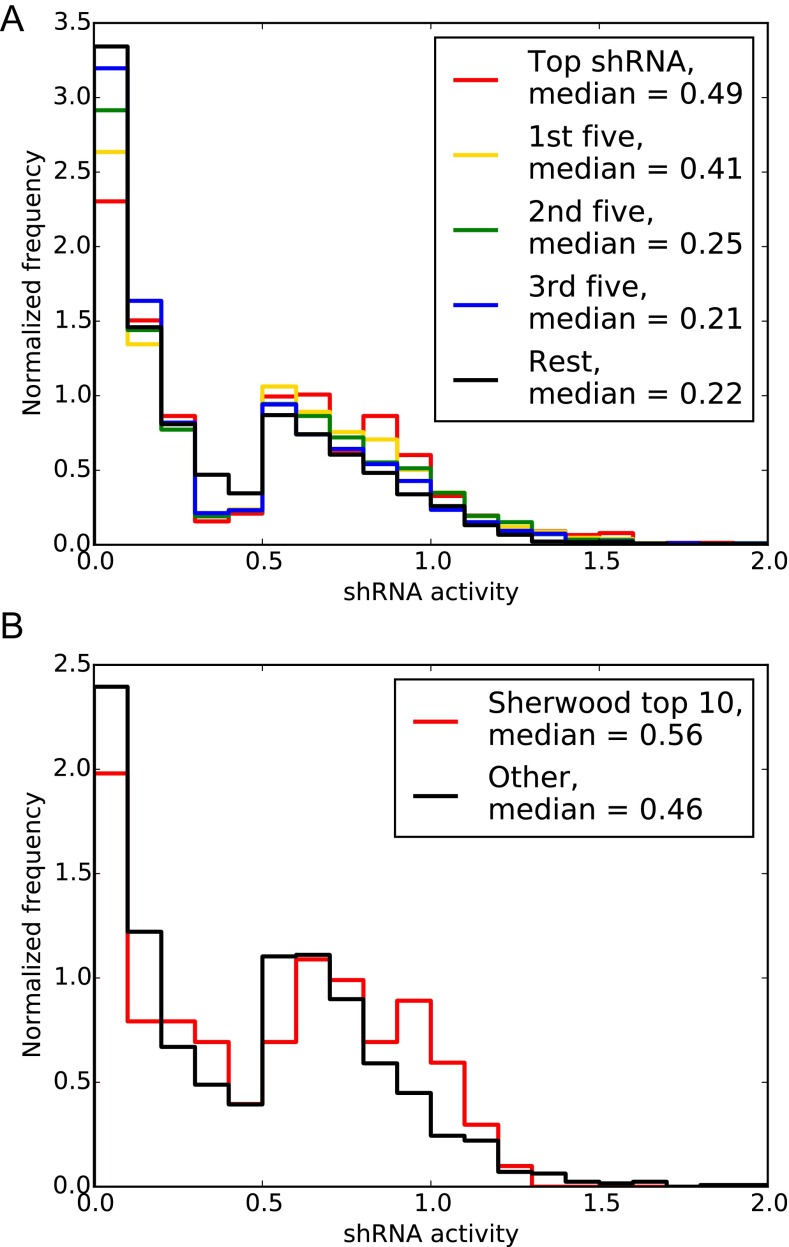
References
-
- Koike-Yusa H, Li Y, Tan EP, Velasco-Herrera MdelC, Yusa K. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat Biotechnol. 2014;32(3):267–273. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
