

Whole-genome sequencing reveals oncogenic mutations in mycosis fungoides
- PMID: 26082451
- PMCID: PMC4513251
- DOI: 10.1182/blood-2014-11-611194
Whole-genome sequencing reveals oncogenic mutations in mycosis fungoides
Abstract
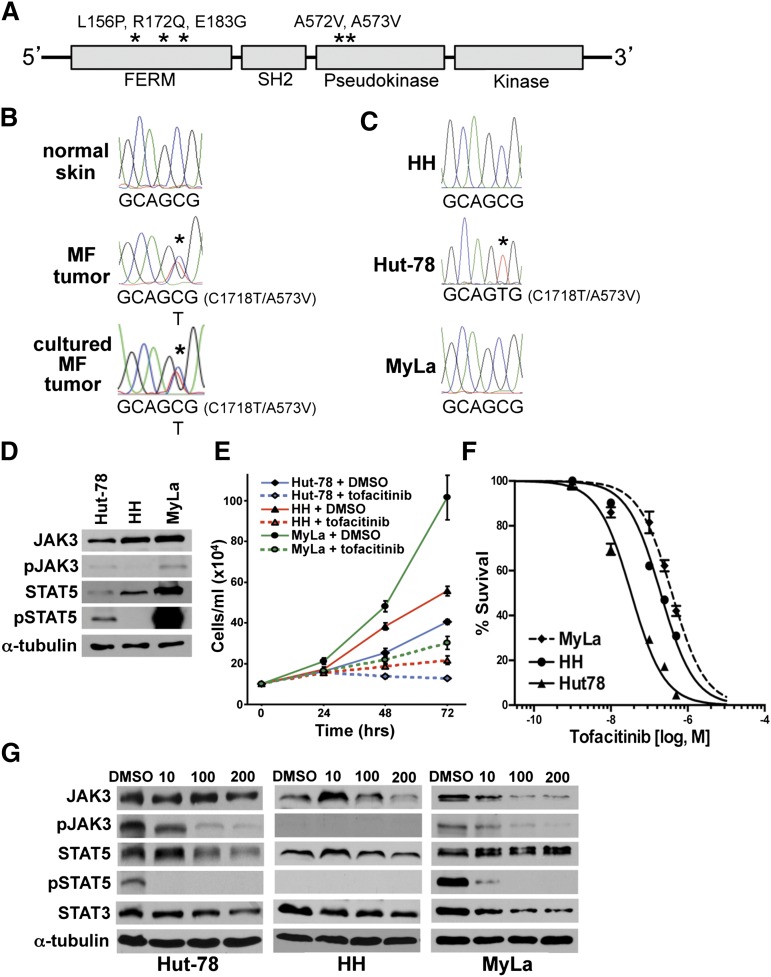
The pathogenesis of mycosis fungoides (MF), the most common cutaneous T-cell lymphoma (CTCL), is unknown. Although genetic alterations have been identified, none are considered consistently causative in MF. To identify potential drivers of MF, we performed whole-genome sequencing of MF tumors and matched normal skin. Targeted ultra-deep sequencing of MF samples and exome sequencing of CTCL cell lines were also performed. Multiple mutations were identified that affected the same pathways, including epigenetic, cell-fate regulation, and cytokine signaling, in MF tumors and CTCL cell lines. Specifically, interleukin-2 signaling pathway mutations, including activating Janus kinase 3 (JAK3) mutations, were detected. Treatment with a JAK3 inhibitor significantly reduced CTCL cell survival. Additionally, the mutation data identified 2 other potential contributing factors to MF, ultraviolet light, and a polymorphism in the tumor suppressor p53 (TP53). Therefore, genetic alterations in specific pathways in MF were identified that may be viable, effective new targets for treatment.
© 2015 by The American Society of Hematology.
Figures
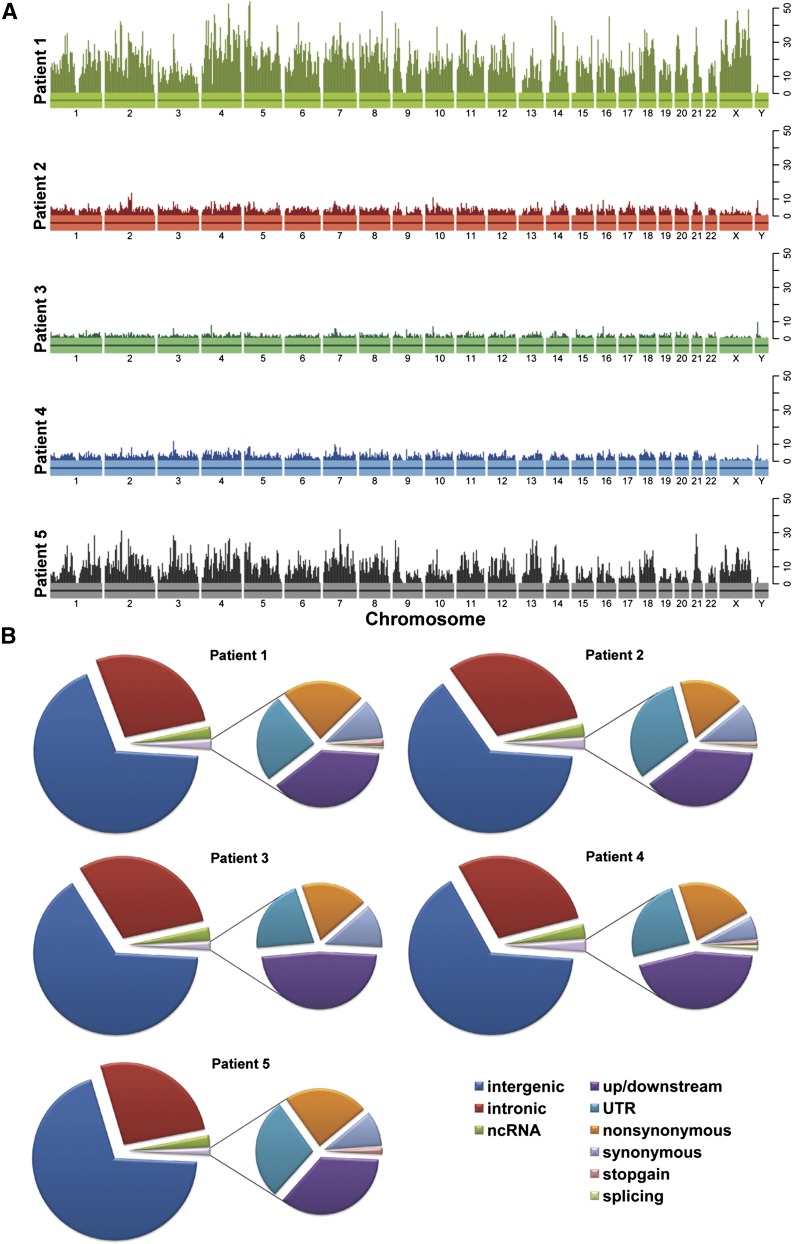
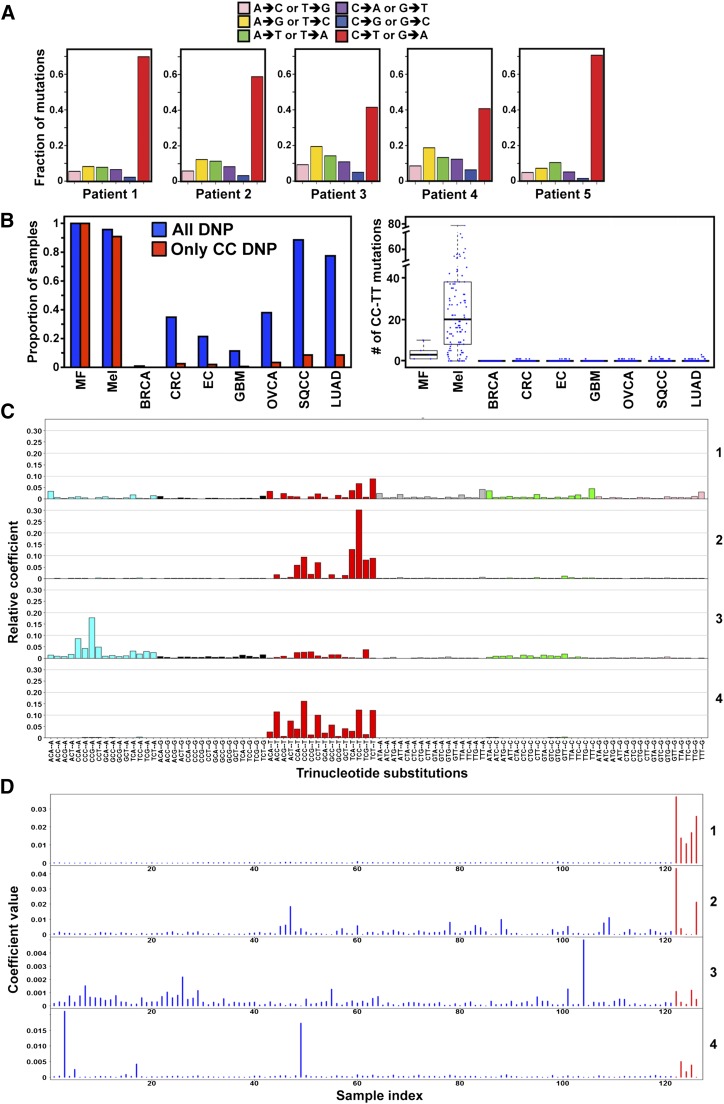
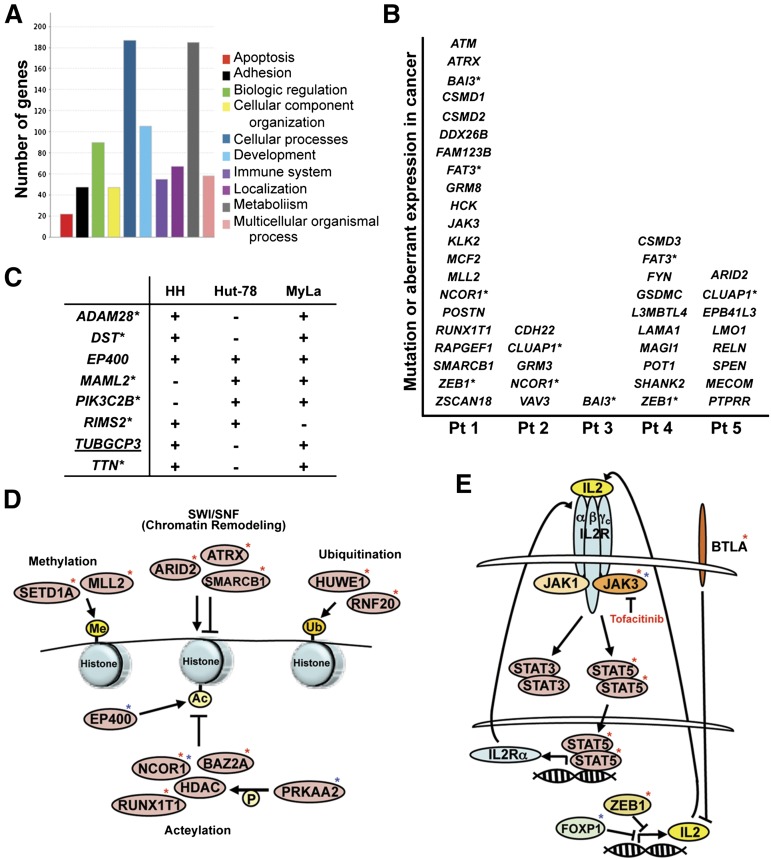
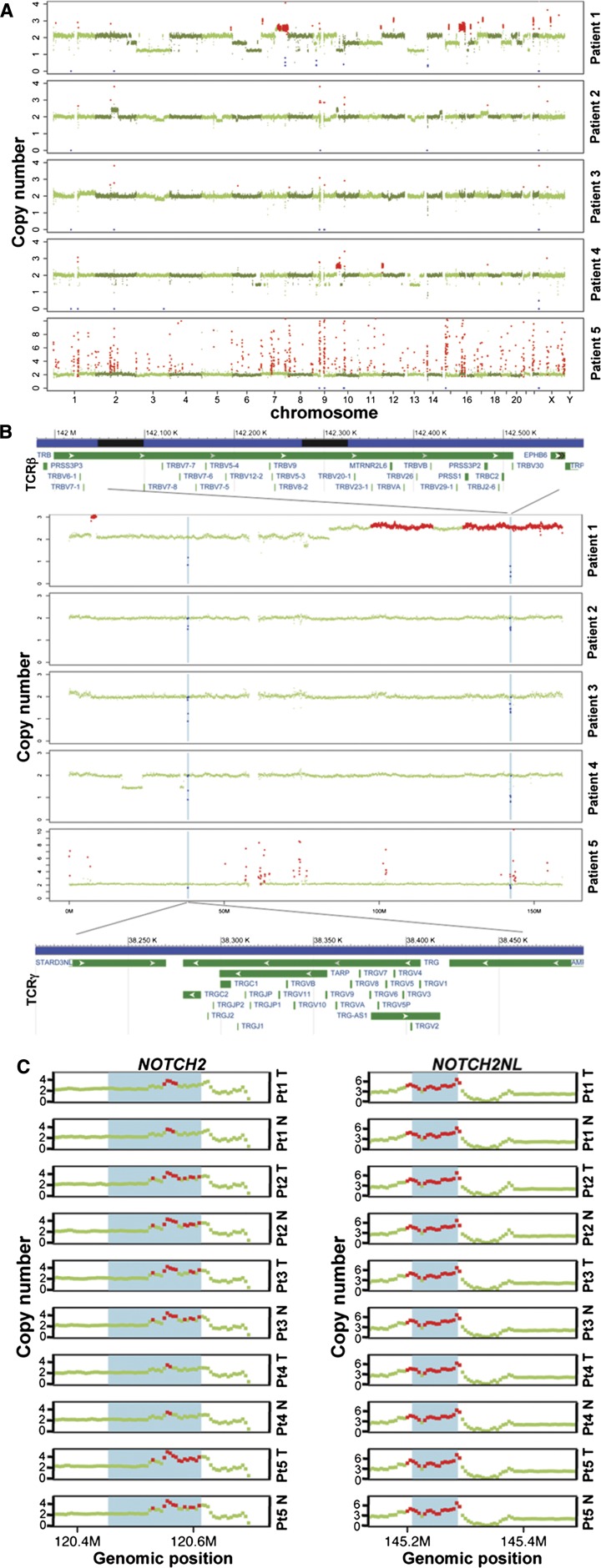
References
-
- Kim YH, Liu HL, Mraz-Gernhard S, Varghese A, Hoppe RT. Long-term outcome of 525 patients with mycosis fungoides and Sezary syndrome: clinical prognostic factors and risk for disease progression. Arch Dermatol. 2003;139(7):857–866. - PubMed
-
- Jackow CM, Cather JC, Hearne V, Asano AT, Musser JM, Duvic M. Association of erythrodermic cutaneous T-cell lymphoma, superantigen-positive Staphylococcus aureus, and oligoclonal T-cell receptor V beta gene expansion. Blood. 1997;89(1):32–40. - PubMed
-
- Tan RS, Butterworth CM, McLaughlin H, Malka S, Samman PD. Mycosis fungoides—a disease of antigen persistence. Br J Dermatol. 1974;91(6):607–616. - PubMed
-
- Wohl Y, Tur E. Environmental risk factors for mycosis fungoides. Curr Probl Dermatol. 2007;35:52–64. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
