

Calculating an optimal box size for ligand docking and virtual screening against experimental and predicted binding pockets
- PMID: 26082804
- PMCID: PMC4468813
- DOI: 10.1186/s13321-015-0067-5
Calculating an optimal box size for ligand docking and virtual screening against experimental and predicted binding pockets
Abstract
Background: Computational approaches have emerged as an instrumental methodology in modern research. For example, virtual screening by molecular docking is routinely used in computer-aided drug discovery. One of the critical parameters for ligand docking is the size of a search space used to identify low-energy binding poses of drug candidates. Currently available docking packages often come with a default protocol for calculating the box size, however, many of these procedures have not been systematically evaluated.
Methods: In this study, we investigate how the docking accuracy of AutoDock Vina is affected by the selection of a search space. We propose a new procedure for calculating the optimal docking box size that maximizes the accuracy of binding pose prediction against a non-redundant and representative dataset of 3,659 protein-ligand complexes selected from the Protein Data Bank. Subsequently, we use the Directory of Useful Decoys, Enhanced to demonstrate that the optimized docking box size also yields an improved ranking in virtual screening. Binding pockets in both datasets are derived from the experimental complex structures and, additionally, predicted by eFindSite.
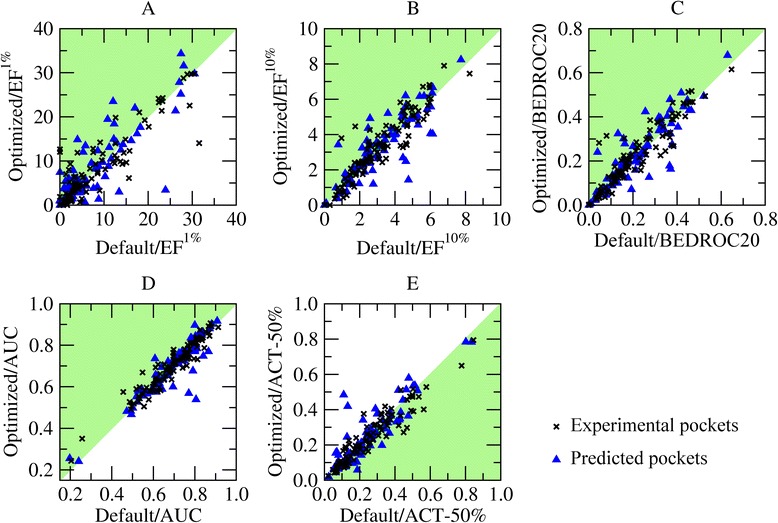
Results: A systematic analysis of ligand binding poses generated by AutoDock Vina shows that the highest accuracy is achieved when the dimensions of the search space are 2.9 times larger than the radius of gyration of a docking compound. Subsequent virtual screening benchmarks demonstrate that this optimized docking box size also improves compound ranking. For instance, using predicted ligand binding sites, the average enrichment factor calculated for the top 1 % (10 %) of the screening library is 8.20 (3.28) for the optimized protocol, compared to 7.67 (3.19) for the default procedure. Depending on the evaluation metric, the optimal docking box size gives better ranking in virtual screening for about two-thirds of target proteins.
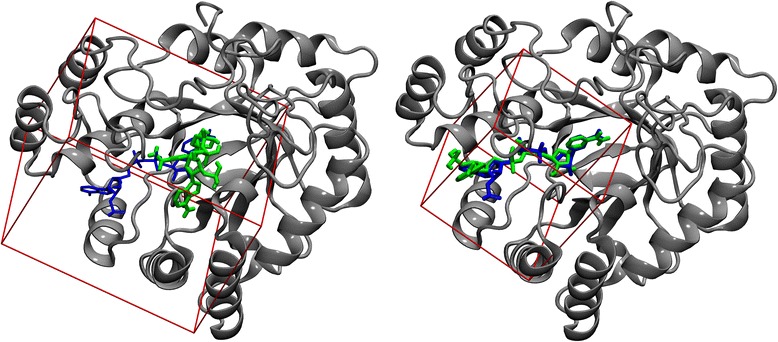
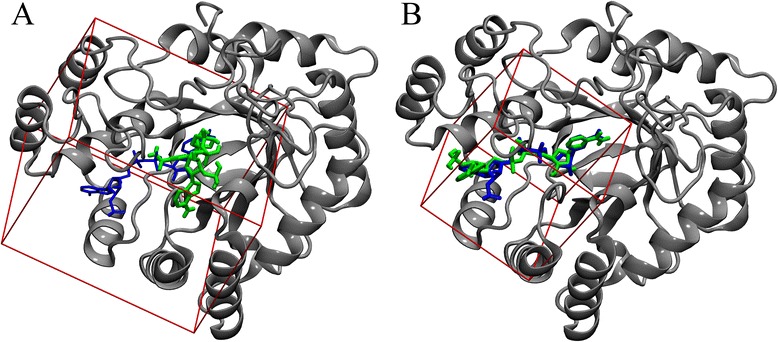
Conclusions: This fully automated procedure can be used to optimize docking protocols in order to improve the ranking accuracy in production virtual screening simulations. Importantly, the optimized search space systematically yields better results than the default method not only for experimental pockets, but also for those predicted from protein structures. A script for calculating the optimal docking box size is freely available at www.brylinski.org/content/docking-box-size. Graphical AbstractWe developed a procedure to optimize the box size in molecular docking calculations. Left panel shows the predicted binding pose of NADP (green sticks) compared to the experimental complex structure of human aldose reductase (blue sticks) using a default protocol. Right panel shows the docking accuracy using an optimized box size.
Keywords: AutoDock Vina; Docking box size; Docking protocols; Ligand binding site prediction; Ligand virtual screening; Molecular docking; Search space.
Figures
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
