

Diversity and Evolution in the Genome of Clostridium difficile
- PMID: 26085550
- PMCID: PMC4475645
- DOI: 10.1128/CMR.00127-14
Diversity and Evolution in the Genome of Clostridium difficile
Abstract
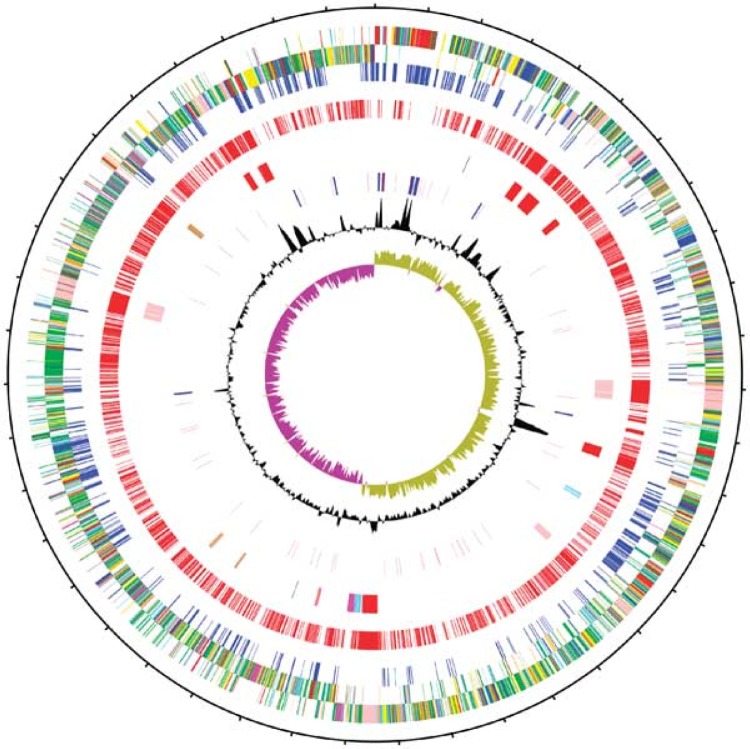
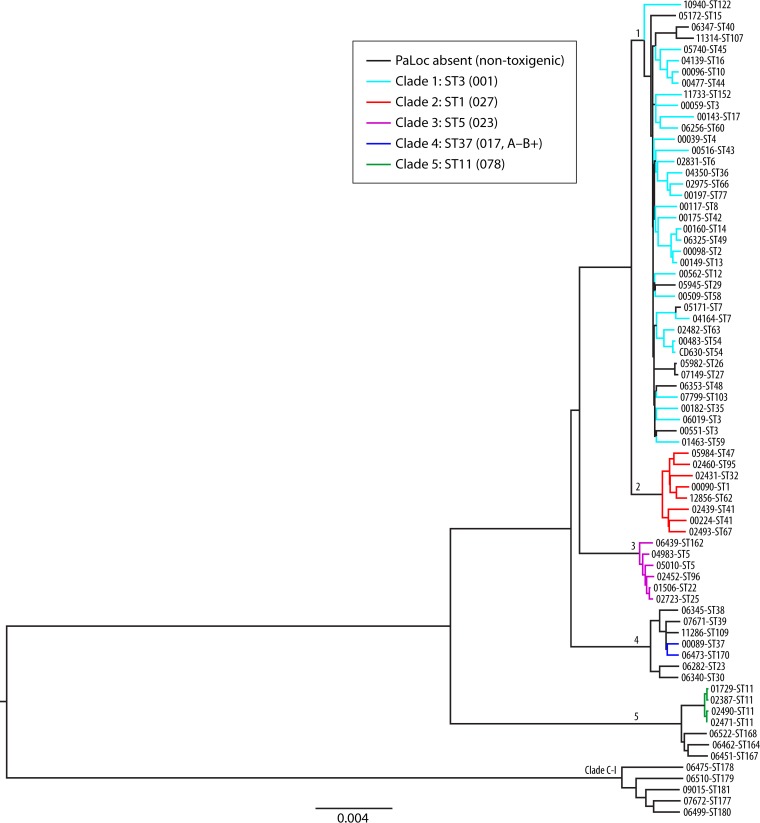
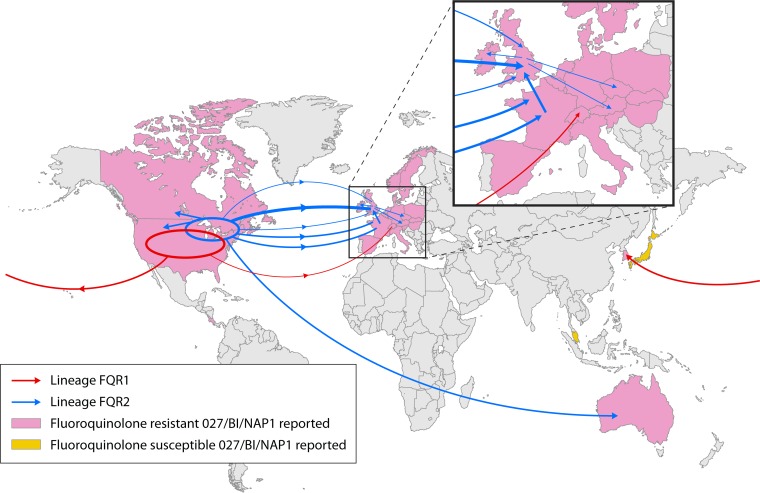
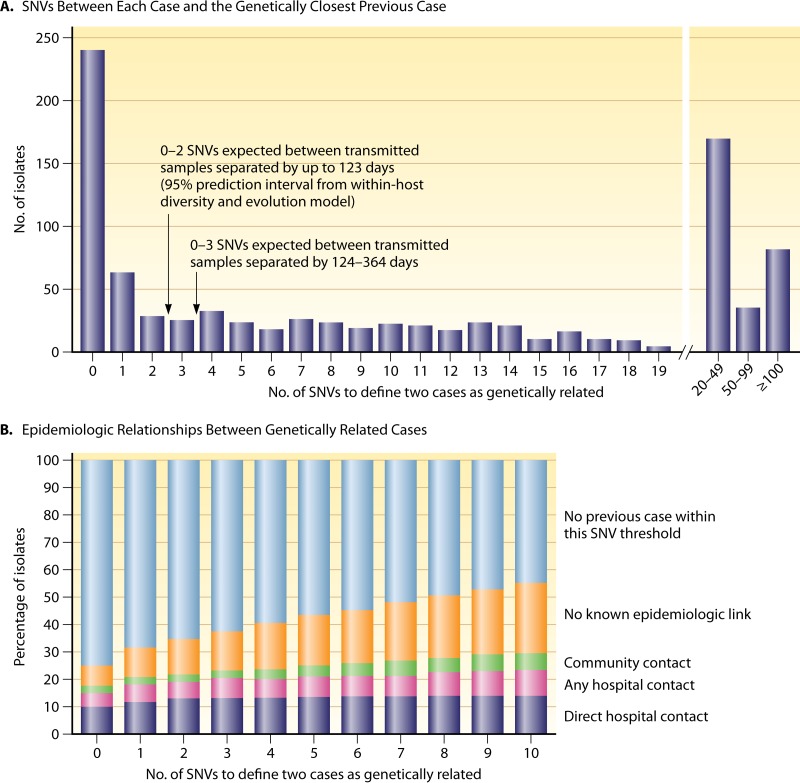
Clostridium difficile infection (CDI) is the leading cause of antimicrobial and health care-associated diarrhea in humans, presenting a significant burden to global health care systems. In the last 2 decades, PCR- and sequence-based techniques, particularly whole-genome sequencing (WGS), have significantly furthered our knowledge of the genetic diversity, evolution, epidemiology, and pathogenicity of this once enigmatic pathogen. C. difficile is taxonomically distinct from many other well-known clostridia, with a diverse population structure comprising hundreds of strain types spread across at least 6 phylogenetic clades. The C. difficile species is defined by a large diverse pangenome with extreme levels of evolutionary plasticity that has been shaped over long time periods by gene flux and recombination, often between divergent lineages. These evolutionary events are in response to environmental and anthropogenic activities and have led to the rapid emergence and worldwide dissemination of virulent clonal lineages. Moreover, genome analysis of large clinically relevant data sets has improved our understanding of CDI outbreaks, transmission, and recurrence. The epidemiology of CDI has changed dramatically over the last 15 years, and CDI may have a foodborne or zoonotic etiology. The WGS era promises to continue to redefine our view of this significant pathogen.
Copyright © 2015, American Society for Microbiology. All Rights Reserved.
Figures





References
-
- Centers for Disease Control and Prevention. 2013. Antibiotic resistance threats in the United States, 2013. Centers for Disease Control and Prevention, Atlanta, GA: http://www.cdc.gov/drugresistance/threat-report-2013/.
-
- Lessa FC, Mu Y, Bamberg WM, Beldavs ZG, Dumyati GK, Dunn JR, Farley MM, Holzbauer SM, Meek JI, Phipps EC, Wilson LE, Winston LG, Cohen JA, Limbago BM, Fridkin SK, Gerding DN, McDonald LC. 2015. Burden of Clostridium difficile infection in the United States. N Engl J Med 372:825–834. doi: 10.1056/NEJMoa1408913. - DOI - PMC - PubMed
-
- Hall IC, O'Toole E. 1935. Intestinal flora in newborn infants with a description of a new pathogenic anaerobe, Bacillus difficilis. Am J Dis Child 49:390–402. doi: 10.1001/archpedi.1935.01970020105010. - DOI
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
