

Epigenomic Signatures of Neuronal Diversity in the Mammalian Brain
- PMID: 26087164
- PMCID: PMC4499463
- DOI: 10.1016/j.neuron.2015.05.018
Epigenomic Signatures of Neuronal Diversity in the Mammalian Brain
Abstract
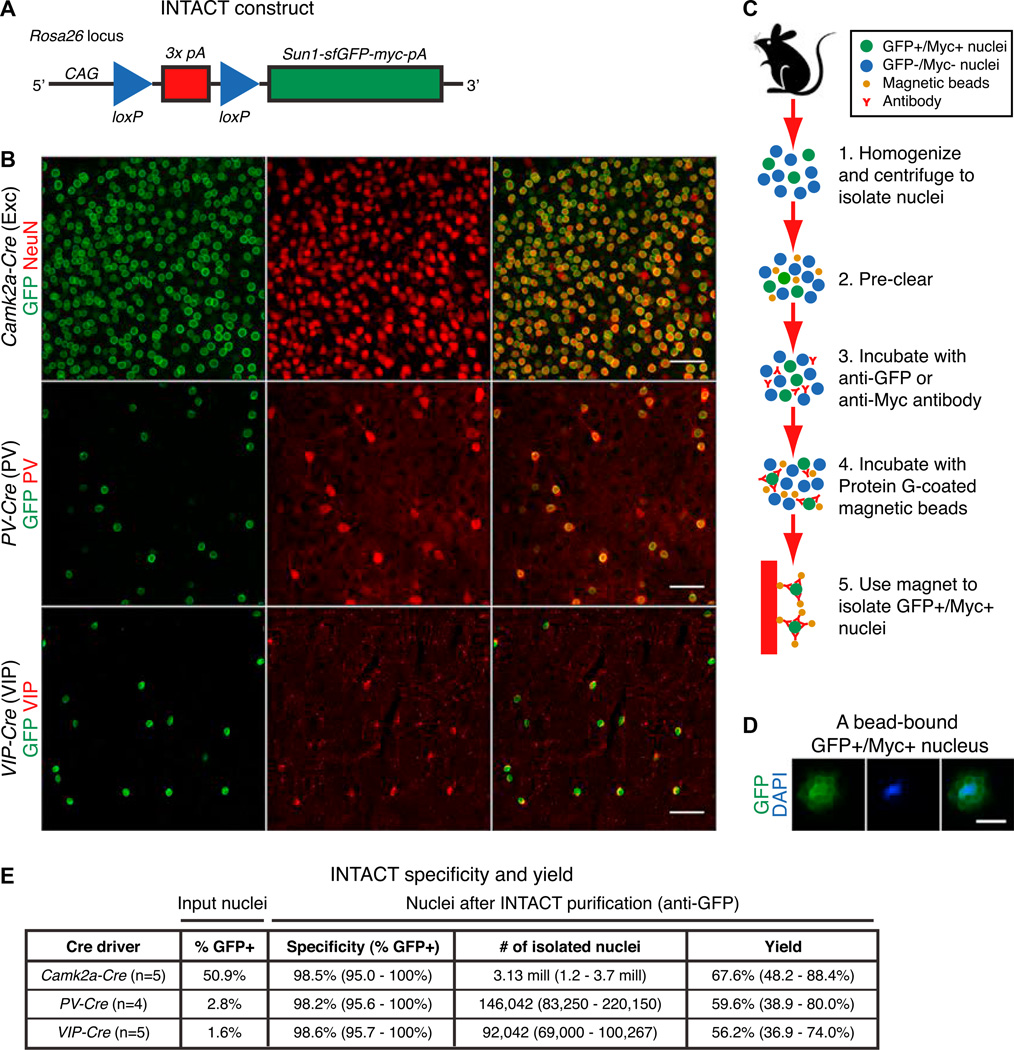
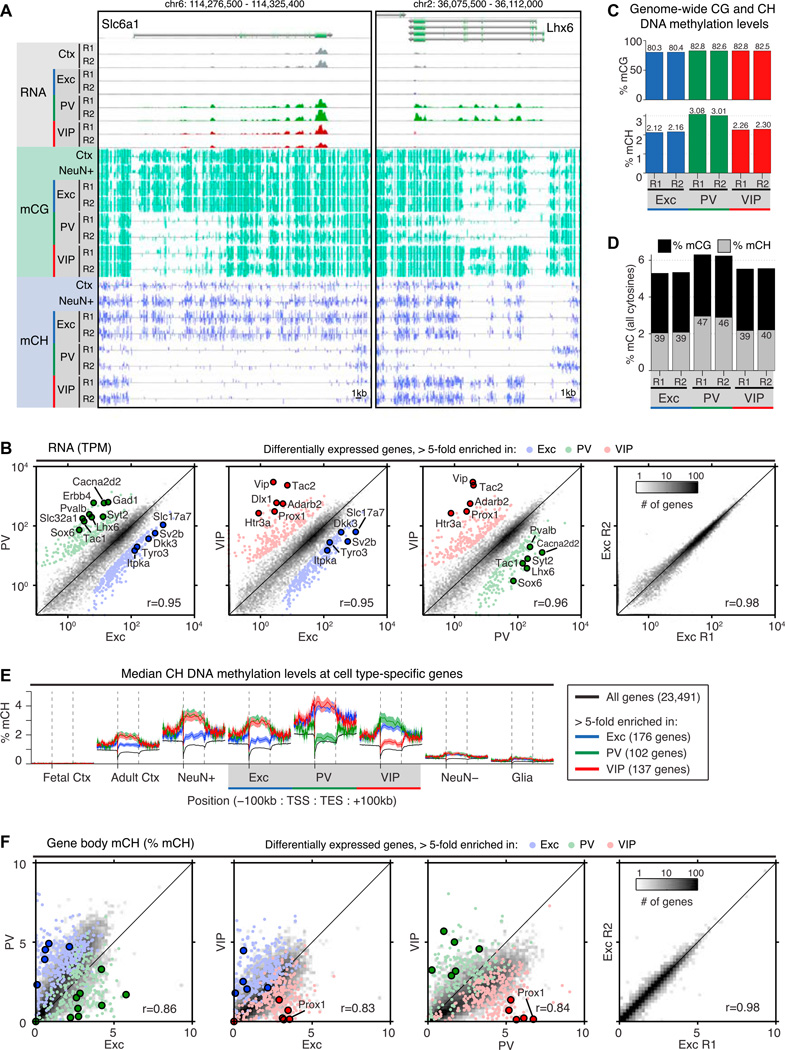
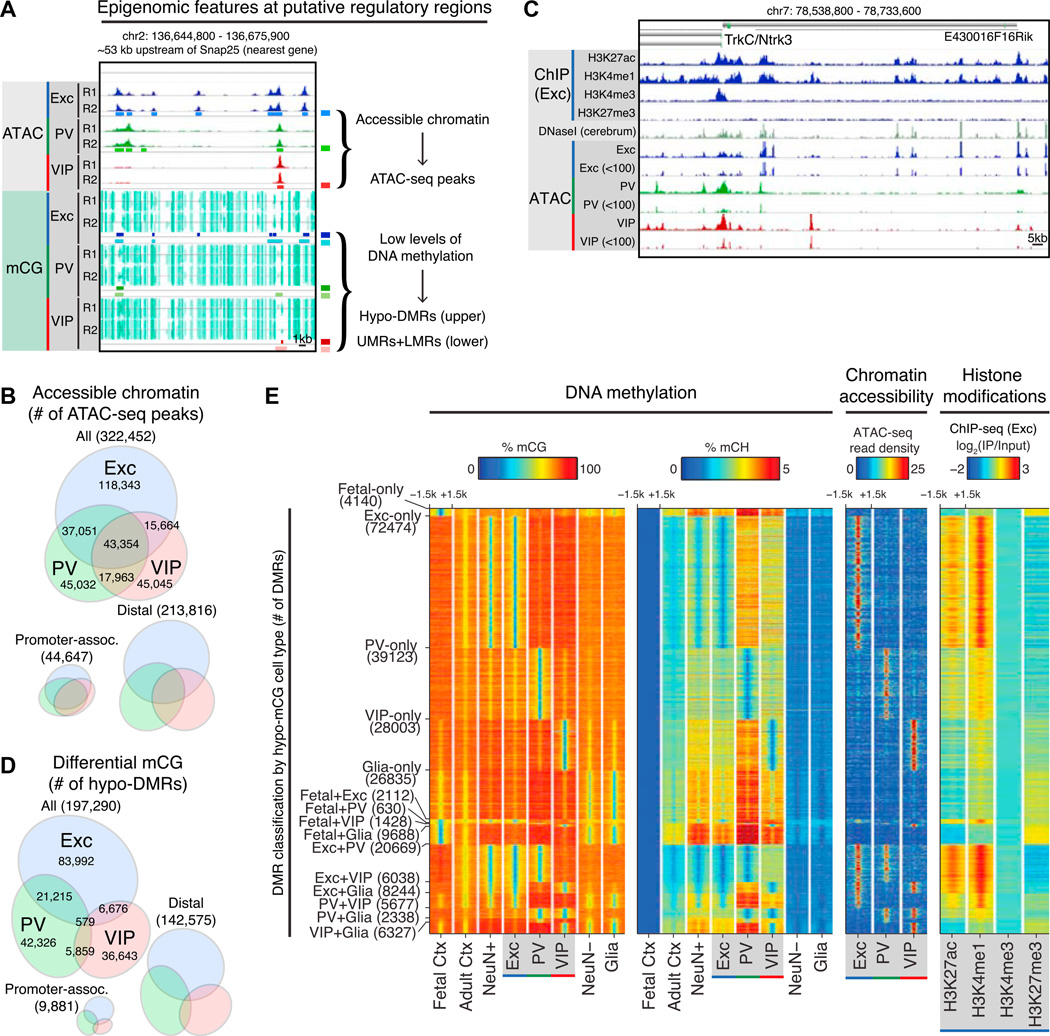
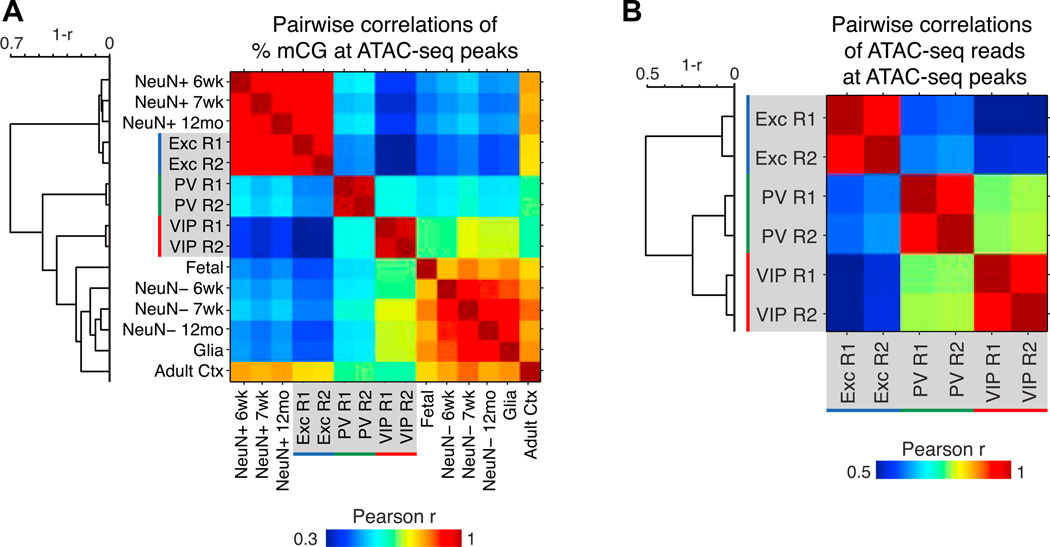
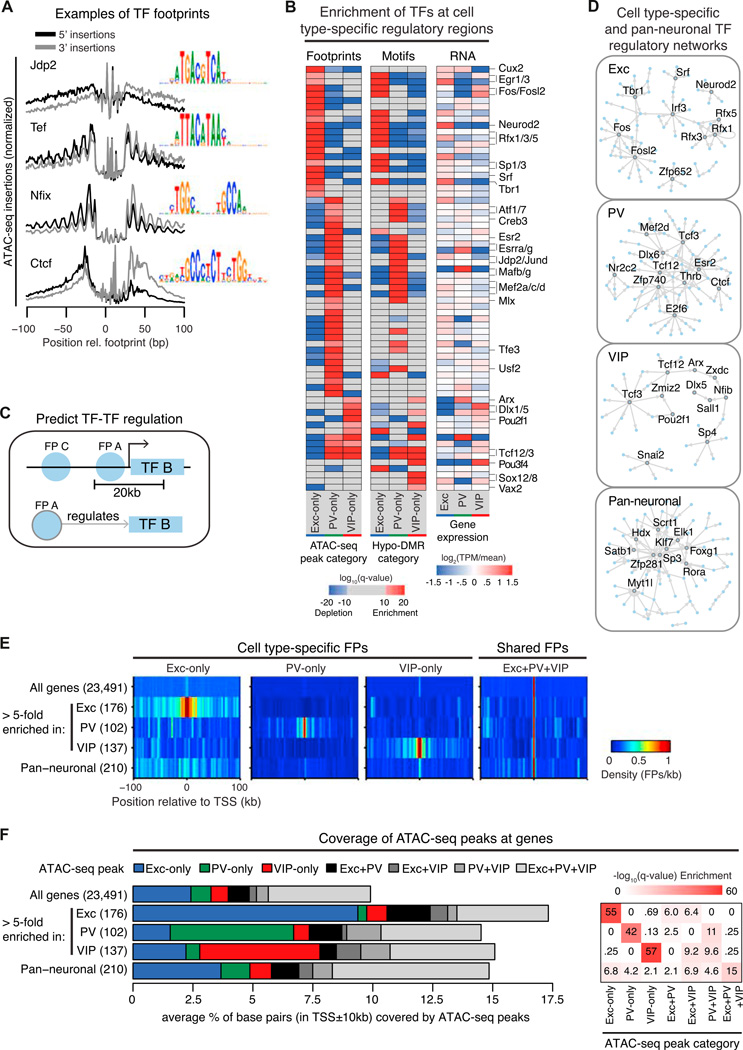
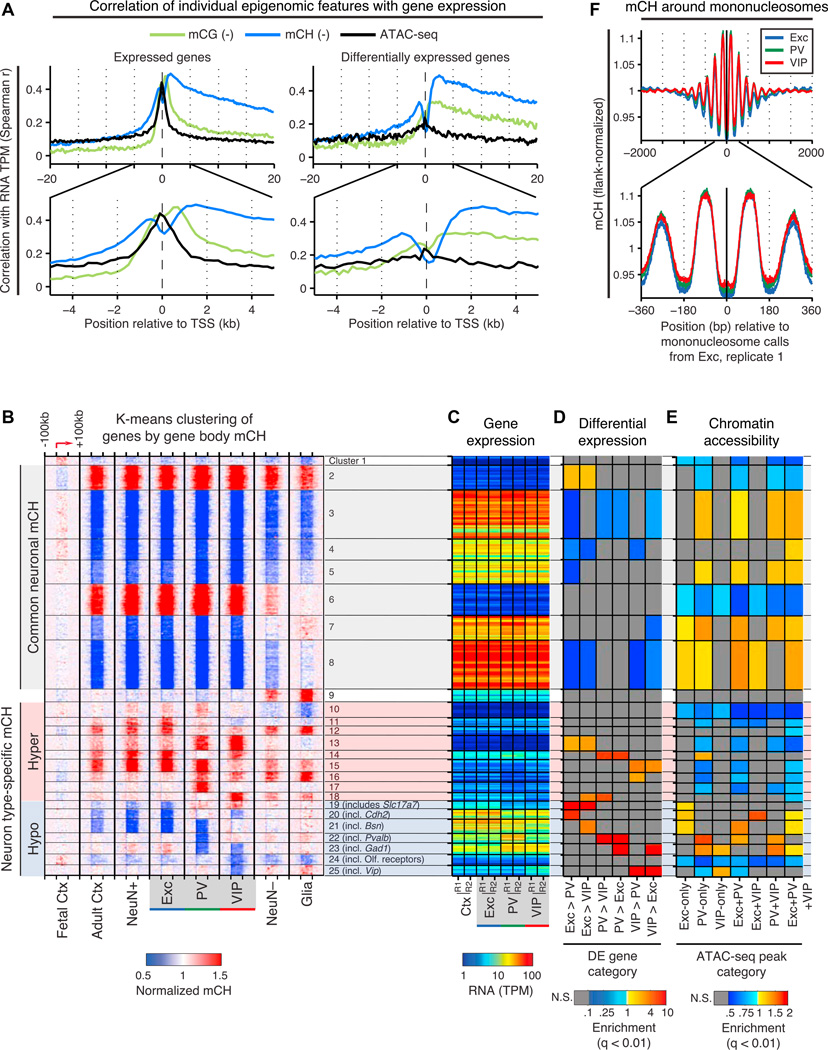
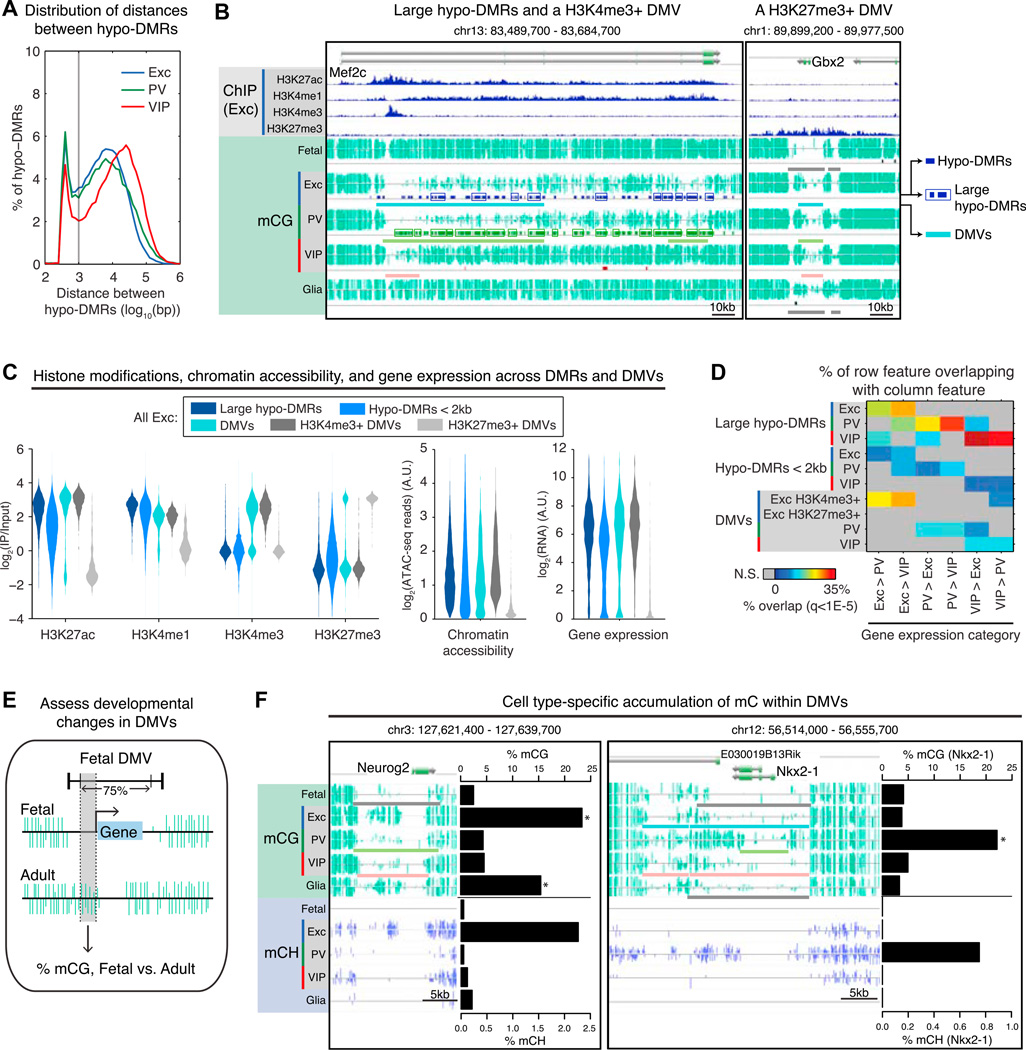
Neuronal diversity is essential for mammalian brain function but poses a challenge to molecular profiling. To address the need for tools that facilitate cell-type-specific epigenomic studies, we developed the first affinity purification approach to isolate nuclei from genetically defined cell types in a mammal. We combine this technique with next-generation sequencing to show that three subtypes of neocortical neurons have highly distinctive epigenomic landscapes. Over 200,000 regions differ in chromatin accessibility and DNA methylation signatures characteristic of gene regulatory regions. By footprinting and motif analyses, these regions are predicted to bind distinct cohorts of neuron subtype-specific transcription factors. Neuronal epigenomes reflect both past and present gene expression, with DNA hyper-methylation at developmentally critical genes appearing as a novel epigenomic signature in mature neurons. Taken together, our findings link the functional and transcriptional complexity of neurons to their underlying epigenomic diversity.
Copyright © 2015 Elsevier Inc. All rights reserved.
Figures
Comment in
-
Epigenomic Landscapes Reflect Neuronal Diversity.Neuron. 2015 Jun 17;86(6):1319-21. doi: 10.1016/j.neuron.2015.06.002. Neuron. 2015. PMID: 26087157
References
-
- Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;76:6–21. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
