

Genetic evidence in the mouse solidifies the calcium hypothesis of myofiber death in muscular dystrophy
- PMID: 26088163
- PMCID: PMC4532779
- DOI: 10.1038/cdd.2015.65
Genetic evidence in the mouse solidifies the calcium hypothesis of myofiber death in muscular dystrophy
Abstract
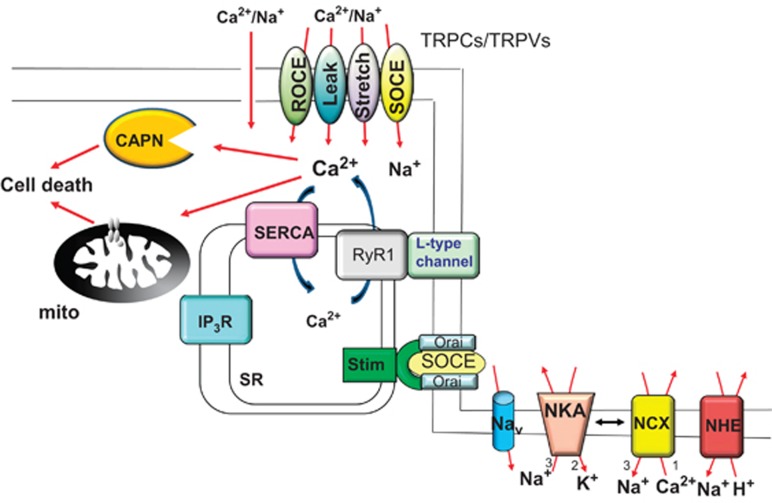
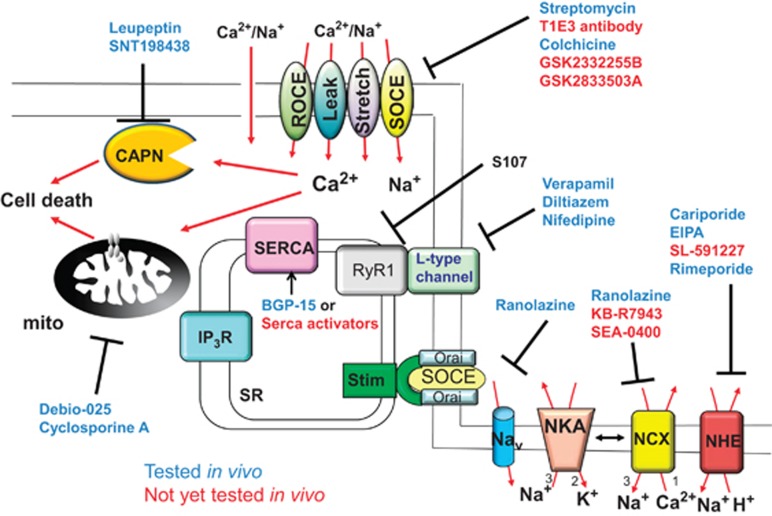
Muscular dystrophy (MD) refers to a clinically and genetically heterogeneous group of degenerative muscle disorders characterized by progressive muscle wasting and often premature death. Although the primary defect underlying most forms of MD typically results from a loss of sarcolemmal integrity, the secondary molecular mechanisms leading to muscle degeneration and myofiber necrosis is debated. One hypothesis suggests that elevated or dysregulated cytosolic calcium is the common transducing event, resulting in myofiber necrosis in MD. Previous measurements of resting calcium levels in myofibers from dystrophic animal models or humans produced equivocal results. However, recent studies in genetically altered mouse models have largely solidified the calcium hypothesis of MD, such that models with artificially elevated calcium in skeletal muscle manifest fulminant dystrophic-like disease, whereas models with enhanced calcium clearance or inhibited calcium influx are resistant to myofiber death and MD. Here, we will review the field and the recent cadre of data from genetically altered mouse models, which we propose have collectively mostly proven the hypothesis that calcium is the primary effector of myofiber necrosis in MD. This new consensus on calcium should guide future selection of drugs to be evaluated in clinical trials as well as gene therapy-based approaches.
Figures
References
-
- Emery AE. Population frequencies of inherited neuromuscular diseases—a world survey. Neuromuscul Dis. 1991;1:19–29. - PubMed
-
- Durbeej M, Campbell KP. Muscular dystrophies involving the dystrophin-glycoprotein complex: an overview of current mouse models. Curr Opin Genet Dev. 2002;12:349–361. - PubMed
-
- Bansal D, Miyake K, Vogel SS, Groh S, Chen CC, Williamson R, et al. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature. 2003;423:168–172. - PubMed
-
- Wrogemann K, Pena SD. Mitochondrial calcium overload: a general mechanism for cell-necrosis in muscle diseases. Lancet. 1976;1:672–674. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
