

Characterization of the Tyrosine Kinase-Regulated Proteome in Breast Cancer by Combined use of RNA interference (RNAi) and Stable Isotope Labeling with Amino Acids in Cell Culture (SILAC) Quantitative Proteomics
- PMID: 26089344
- PMCID: PMC4563730
- DOI: 10.1074/mcp.M115.048090
Characterization of the Tyrosine Kinase-Regulated Proteome in Breast Cancer by Combined use of RNA interference (RNAi) and Stable Isotope Labeling with Amino Acids in Cell Culture (SILAC) Quantitative Proteomics
Abstract
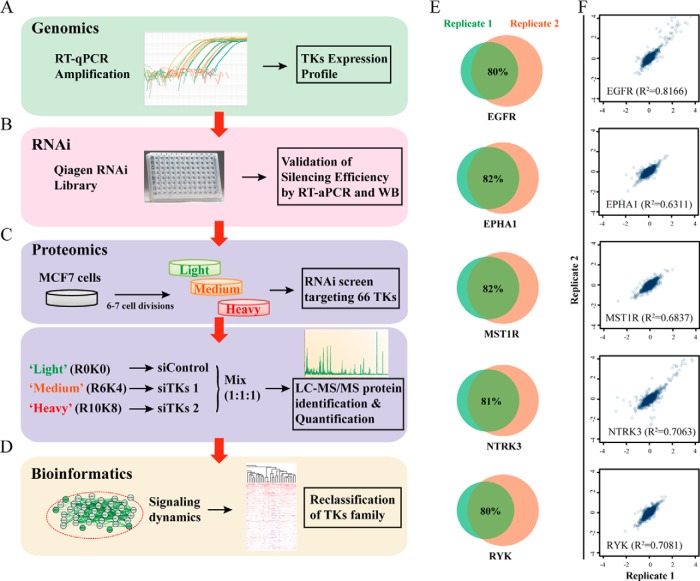
Tyrosine kinases (TKs) are central regulators in cellular activities and perturbations of TK signaling contribute to oncogenesis. However, less than half of the TKs have been thoroughly studied and a global functional analysis of their proteomic portrait is lacking. Here we conducted a combined approach of RNA interference (RNAi) and stable isotope labeling with amino acids in cell culture (SILAC)-based quantitative proteomics to decode the TK-regulated proteome and associated signaling dynamics. As a result, a broad proteomic repertoire modulated by TKs was revealed, upon silencing of the 65 TKs expressed in MCF7 breast cancer cells. This yielded 10 new distinctive TK clusters according to similarity in TK-regulated proteome, each characterized by a unique signaling signature in contrast to previous classifications. We provide functional analyses and identify critical pathways for each cluster based on their common downstream targets. Analysis of different breast cancer subtypes showed distinct correlations of each cluster with clinical outcome. From the significantly up- and down-regulated proteins, we identified a number of markers of drug sensitivity and resistance. These data supports the role of TKs in regulating major aspects of cellular activity, but also reveals redundancy in signaling, explaining why kinase inhibitors alone often fail to achieve their clinical aims. The TK-SILACepedia provides a comprehensive resource for studying the global function of TKs in cancer.
© 2015 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures






References
-
- Hanahan D., Weinberg R. A. (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 - PubMed
-
- Kolch W., Pitt A. (2010) Functional proteomics to dissect tyrosine kinase signalling pathways in cancer. Nat. Rev. Cancer 10, 618–629 - PubMed
-
- Gstaiger M., Aebersold R. (2009) Applying mass spectrometry-based proteomics to genetics, genomics, and network biology. Nat. Rev. Genet. 10, 617–627 - PubMed
-
- Amanchy R., Kalume D. E., Iwahori A., Zhong J., Pandey A. (2005) Phosphoproteome analysis of HeLa cells using stable isotope labeling with amino acids in cell culture (SILAC). J. Proteome Res. 4, 1661–1671 - PubMed
-
- Olsen J. V., Blagoev B., Gnad F., Macek B., Kumar C., Mortensen P., Mann M. (2006) Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–648 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
