

Loss of tau rescues inflammation-mediated neurodegeneration
- PMID: 26089772
- PMCID: PMC4452825
- DOI: 10.3389/fnins.2015.00196
Loss of tau rescues inflammation-mediated neurodegeneration
Abstract
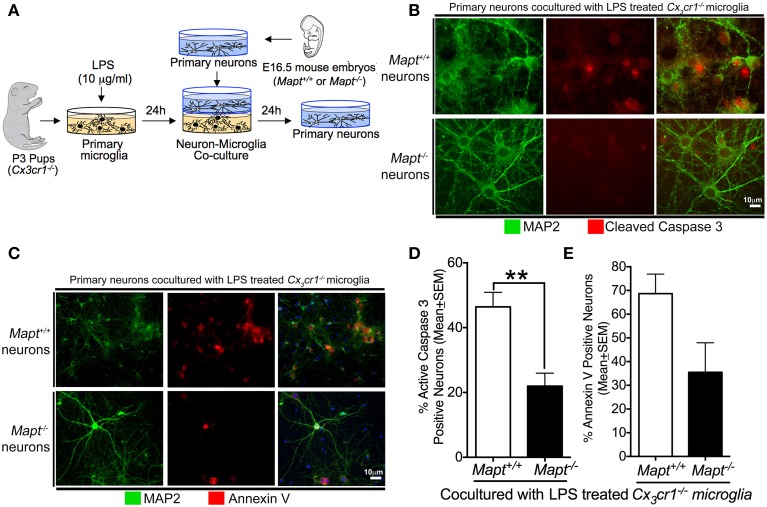
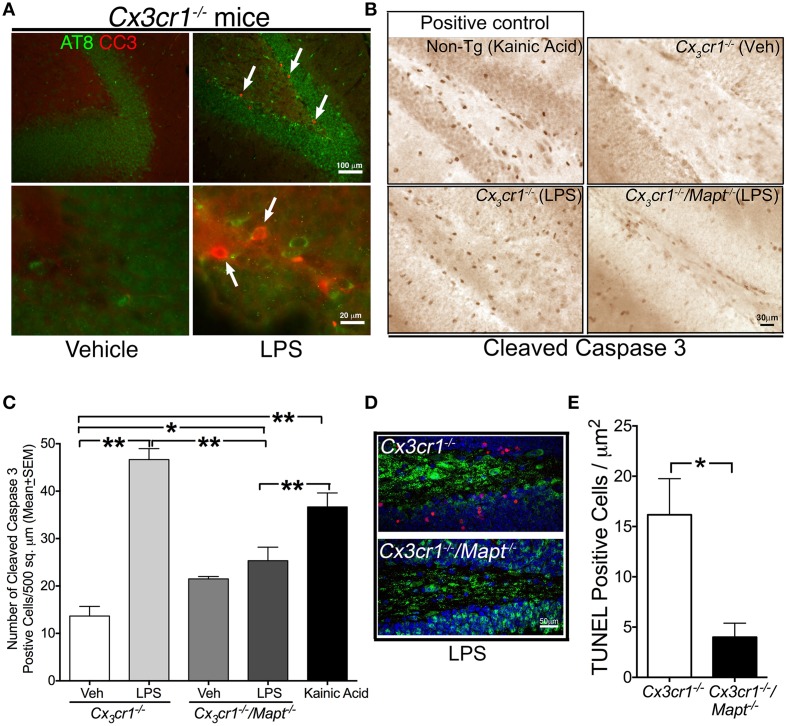
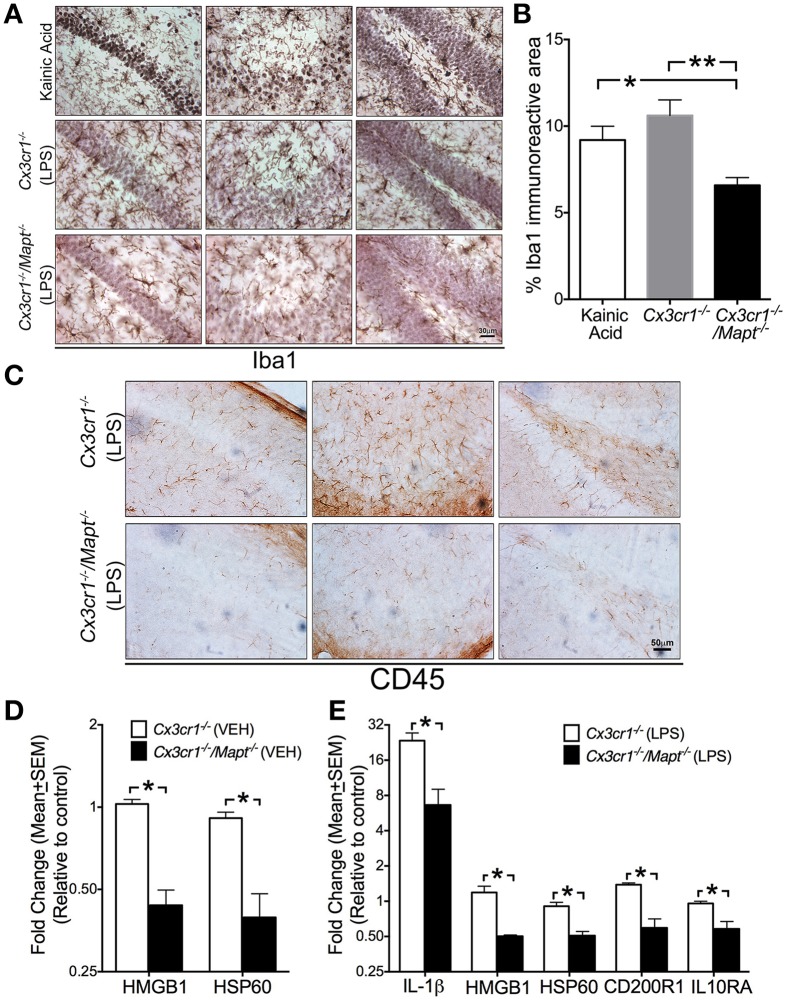
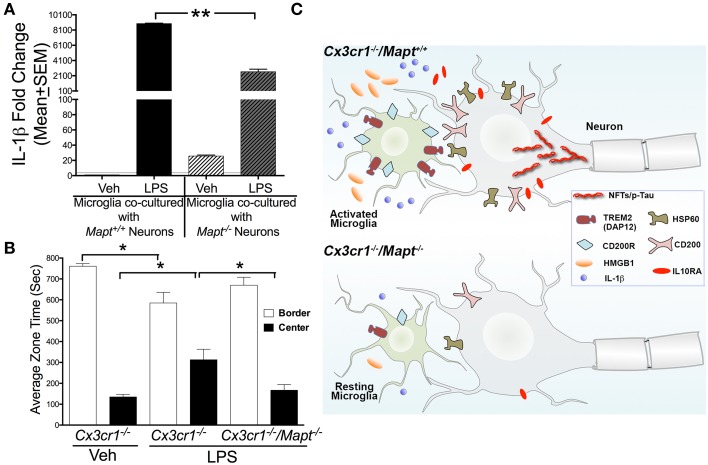
Neuroinflammation is one of the neuropathological hallmarks of Alzheimer's disease (AD) and related tauopathies. Activated microglia spatially coexist with microtubule-associated protein tau (Mapt or tau)-burdened neurons in the brains of human AD and non-AD tauopathies. Numerous studies have suggested that neuroinflammation precedes tau pathology and that induction or blockage of neuroinflammation via lipopolysaccharide (LPS) or anti-inflammatory compounds (such as FK506) accelerate or block tau pathology, respectively in several animal models of tauopathy. We have previously demonstrated that microglia-mediated neuroinflammation via deficiency of the microglia-specific chemokine (fractalkine) receptor, CX3CR1, promotes tau pathology and neurodegeneration in a mouse model of LPS-induced systemic inflammation. Here, we demonstrate that tau mediates the neurotoxic effects of LPS in Cx3cr1 (-/-) mice. First, Mapt (+/+) neurons displayed elevated levels of Annexin V (A5) and TUNEL (markers of neurodegeneration) when co-cultured with LPS-treated Cx3cr1 (-/-)microglia, which is rescued in Mapt (-/-) neurons. Second, a neuronal population positive for phospho-S199 (AT8) tau in the dentate gyrus is also positive for activated or cleaved caspase (CC3) in the LPS-treated Cx3cr1 (-/-) mice. Third, genetic deficiency for tau in Cx3cr1 (-/-) mice resulted in reduced microglial activation, altered expression of inflammatory genes and a significant reduction in the number of neurons positive for CC3 compared to Cx3cr1 (-/-)mice. Finally, Cx3cr1 (-/-)mice exposed to LPS displayed a lack of inhibition in an open field exploratory behavioral test, which is rescued by tau deficiency. Taken together, our results suggest that pathological alterations in tau mediate inflammation-induced neurotoxicity and that deficiency of Mapt is neuroprotective. Thus, therapeutic approaches toward either reducing tau levels or blocking neuroinflammatory pathways may serve as a potential strategy in treating tauopathies.
Keywords: Alzheimer's disease; CX3CR1; microglia; microtubule associated protein tau (MAPT); neurodegeneration; neuroinflammation; tau protein; tauopathies.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials