

Paired-Duplication Signatures Mark Cryptic Inversions and Other Complex Structural Variation
- PMID: 26094575
- PMCID: PMC4571023
- DOI: 10.1016/j.ajhg.2015.05.012
Paired-Duplication Signatures Mark Cryptic Inversions and Other Complex Structural Variation
Abstract
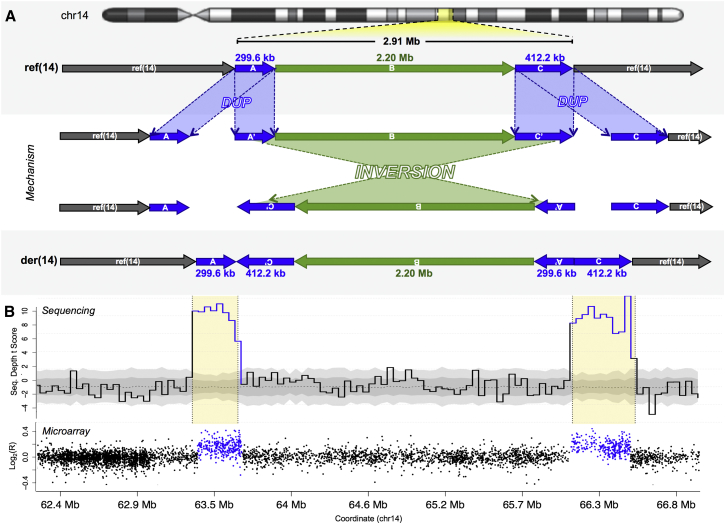
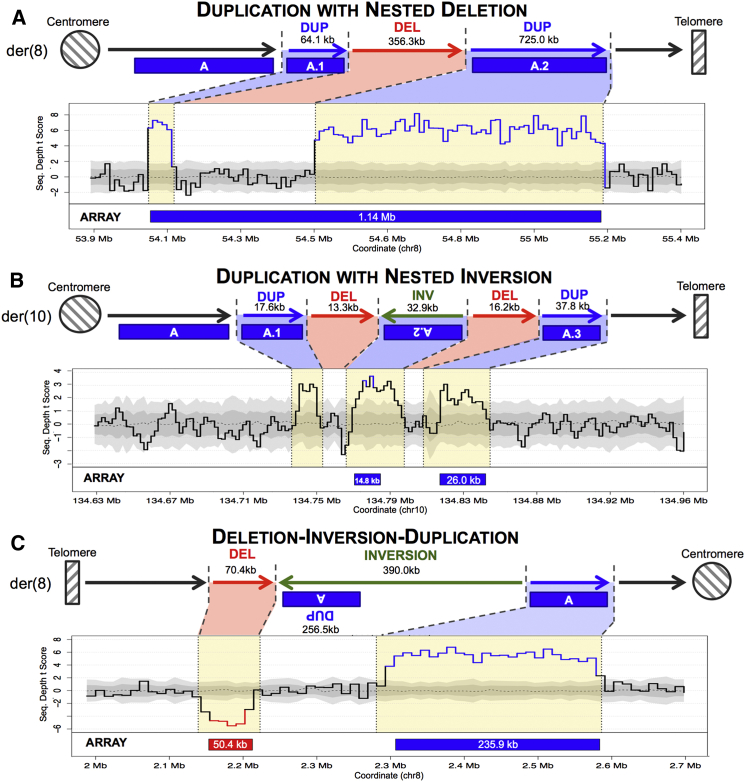
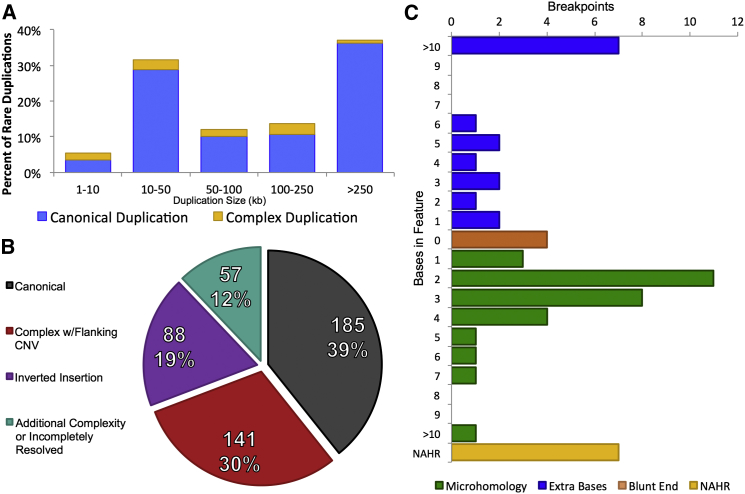
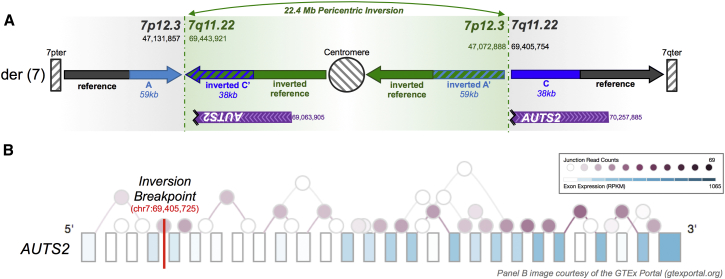
Copy-number variants (CNVs) have been the predominant focus of genetic studies of structural variation, and chromosomal microarray (CMA) for genome-wide CNV detection is the recommended first-tier genetic diagnostic screen in neurodevelopmental disorders. We compared CNVs observed by CMA to the structural variation detected by whole-genome large-insert sequencing in 259 individuals diagnosed with autism spectrum disorder (ASD) from the Simons Simplex Collection. These analyses revealed a diverse landscape of complex duplications in the human genome. One remarkably common class of complex rearrangement, which we term dupINVdup, involves two closely located duplications ("paired duplications") that flank the breakpoints of an inversion. This complex variant class is cryptic to CMA, but we observed it in 8.1% of all subjects. We also detected other paired-duplication signatures and duplication-mediated complex rearrangements in 15.8% of all ASD subjects. Breakpoint analysis showed that the predominant mechanism of formation of these complex duplication-associated variants was microhomology-mediated repair. On the basis of the striking prevalence of dupINVdups in this cohort, we explored the landscape of all inversion variation among the 235 highest-quality libraries and found abundant complexity among these variants: only 39.3% of inversions were canonical, or simple, inversions without additional rearrangement. Collectively, these findings indicate that dupINVdups, as well as other complex duplication-associated rearrangements, represent relatively common sources of genomic variation that is cryptic to population-based microarray and low-depth whole-genome sequencing. They also suggest that paired-duplication signatures detected by CMA warrant further scrutiny in genetic diagnostic testing given that they might mark complex rearrangements of potential clinical relevance.
Copyright © 2015 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
