

Outcome of the First wwPDB Hybrid/Integrative Methods Task Force Workshop
- PMID: 26095030
- PMCID: PMC4933300
- DOI: 10.1016/j.str.2015.05.013
Outcome of the First wwPDB Hybrid/Integrative Methods Task Force Workshop
Abstract
Structures of biomolecular systems are increasingly computed by integrative modeling that relies on varied types of experimental data and theoretical information. We describe here the proceedings and conclusions from the first wwPDB Hybrid/Integrative Methods Task Force Workshop held at the European Bioinformatics Institute in Hinxton, UK, on October 6 and 7, 2014. At the workshop, experts in various experimental fields of structural biology, experts in integrative modeling and visualization, and experts in data archiving addressed a series of questions central to the future of structural biology. How should integrative models be represented? How should the data and integrative models be validated? What data should be archived? How should the data and models be archived? What information should accompany the publication of integrative models?
Copyright © 2015 Elsevier Ltd. All rights reserved.
Figures
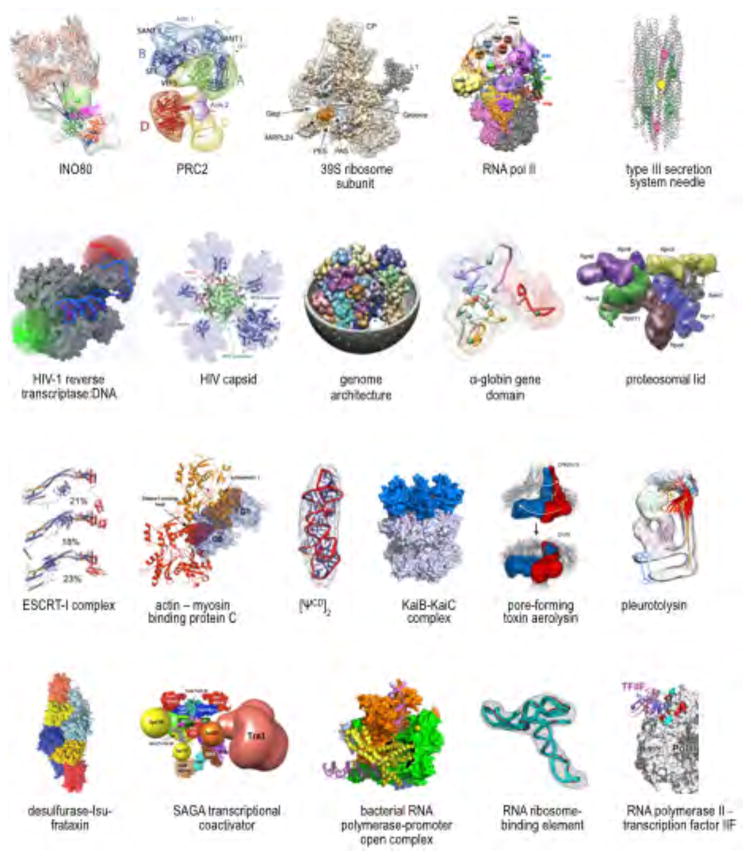
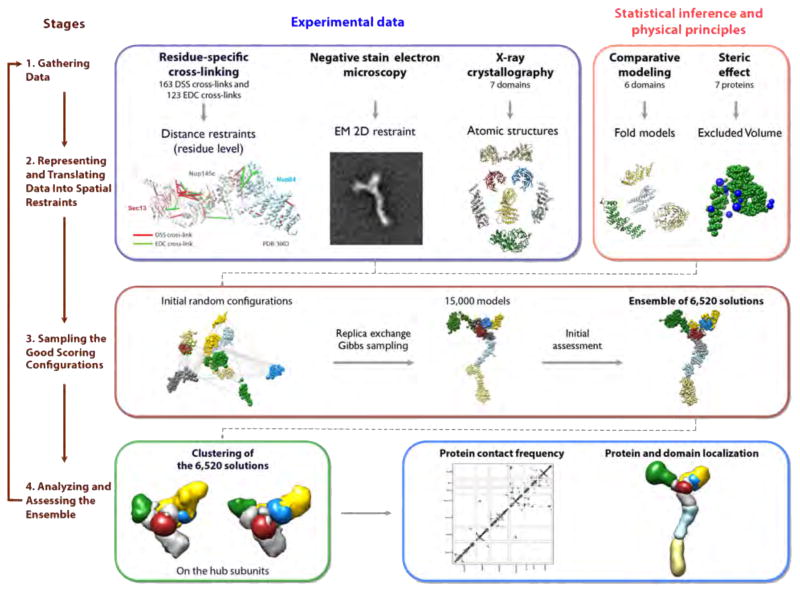
References
-
- Alber F, Dokudovskaya S, Veenhoff L, Zhang W, Kipper J, Devos D, Suprapto A, Karni-Schmidt O, Williams R, Chait B, et al. Determining the architectures of macromolecular assemblies. Nature. 2007;450:683–694. - PubMed
-
- Alber F, Forster F, Korkin D, Topf M, Sali A. Integrating diverse data for structure determination of macromolecular assemblies. Annu Rev Biochem. 2008;77:443–477. - PubMed
-
- Ando T. High-speed AFM imaging. Curr Opin Struct Biol. 2014;28:63–68. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- P41 LM05799/LM/NLM NIH HHS/United States
- P01 AG002132/AG/NIA NIH HHS/United States
- 082961/WT_/Wellcome Trust/United Kingdom
- P41 LM005799/LM/NLM NIH HHS/United States
- R01 GM079429/GM/NIGMS NIH HHS/United States
- GM079429/GM/NIGMS NIH HHS/United States
- 088944/WT_/Wellcome Trust/United Kingdom
- 103139/Z/13/Z/WT_/Wellcome Trust/United Kingdom
- 103139/WT_/Wellcome Trust/United Kingdom
- U01 GM093324/GM/NIGMS NIH HHS/United States
- 100140/WT_/Wellcome Trust/United Kingdom
- MC_U105184322/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
