

A Conserved Circular Network of Coregulated Lipids Modulates Innate Immune Responses
- PMID: 26095250
- PMCID: PMC4523684
- DOI: 10.1016/j.cell.2015.05.051
A Conserved Circular Network of Coregulated Lipids Modulates Innate Immune Responses
Abstract
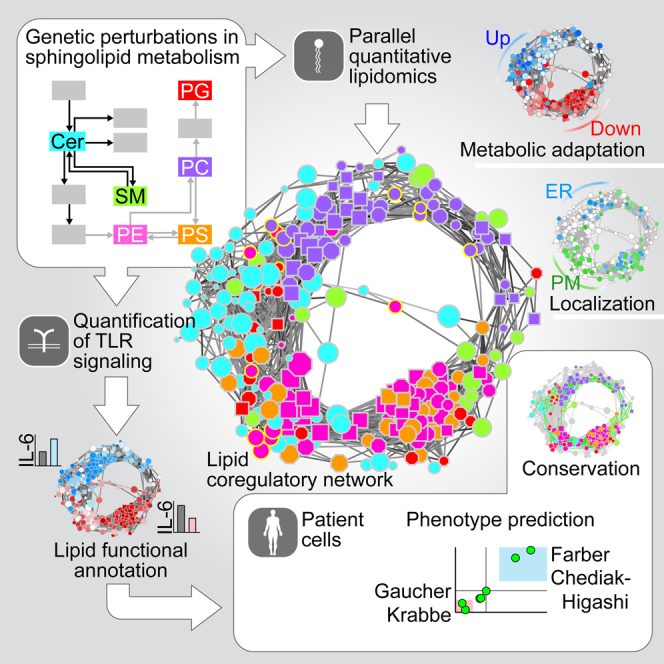
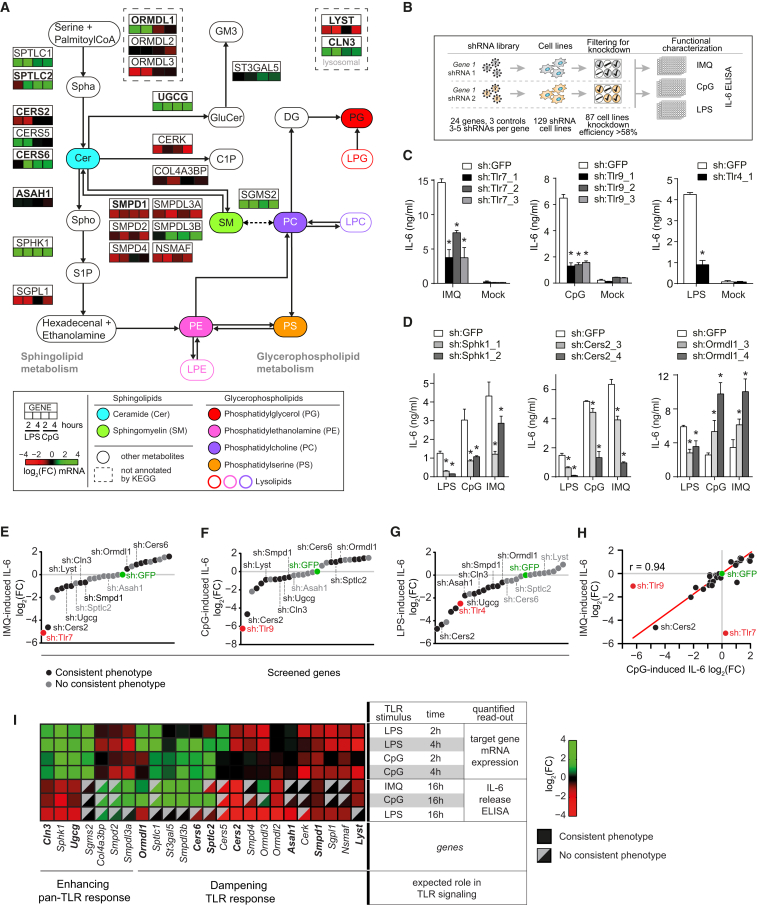
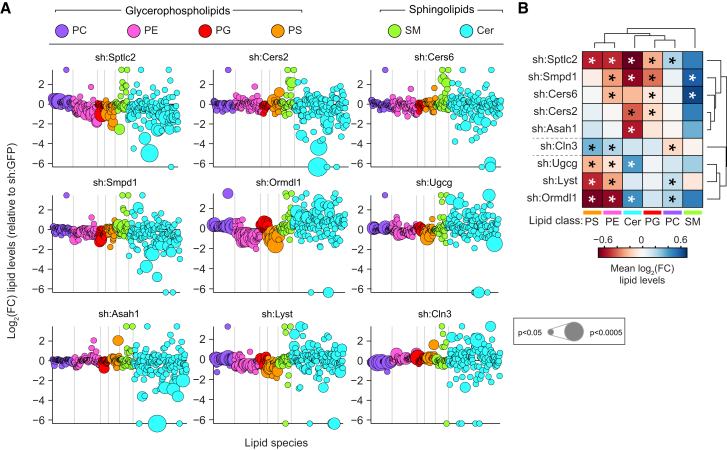
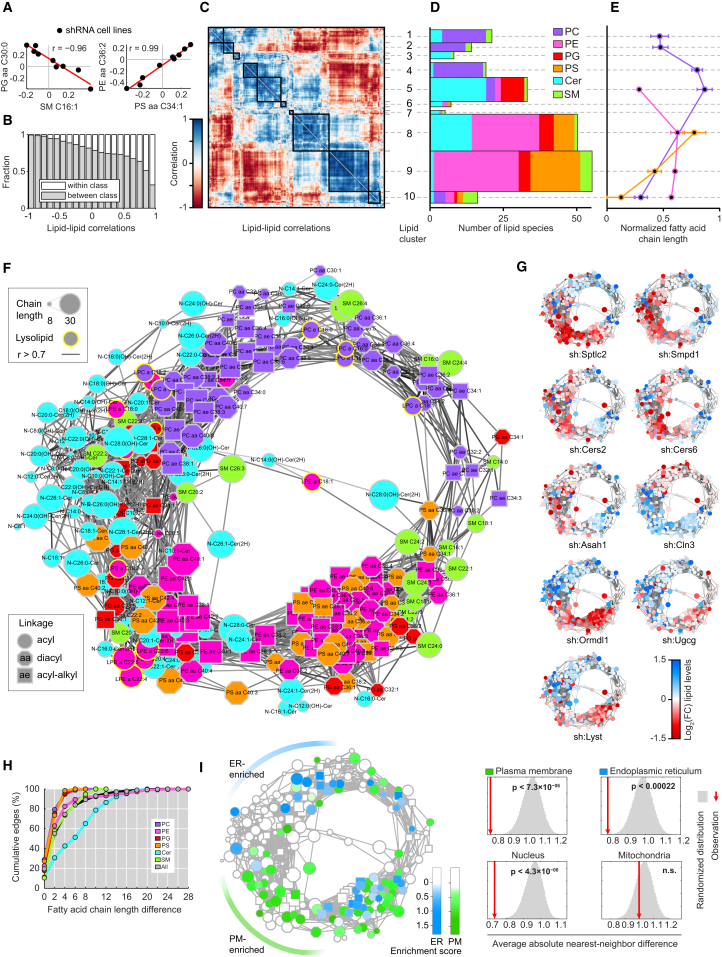
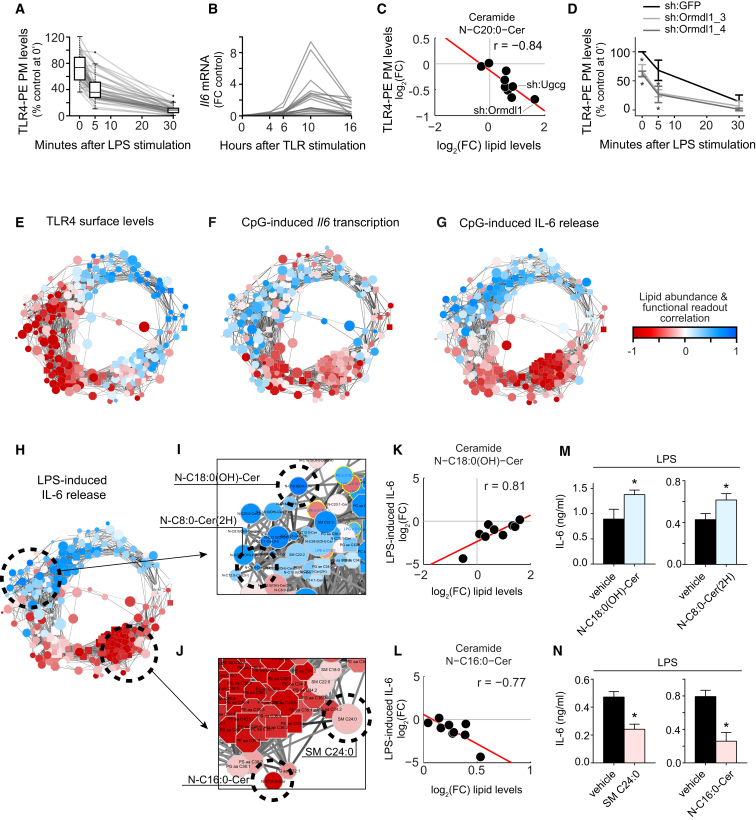
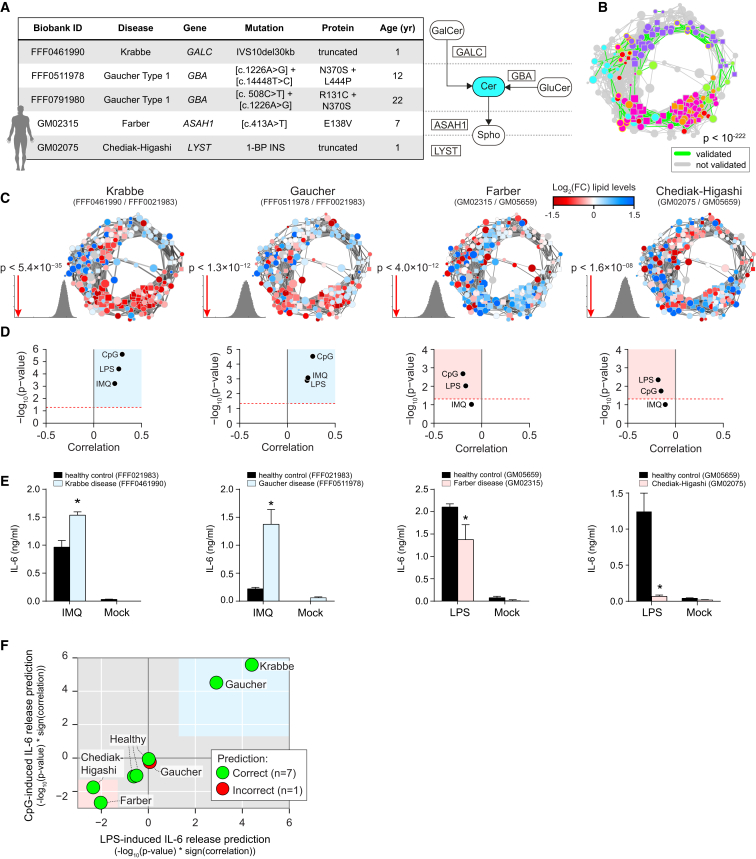
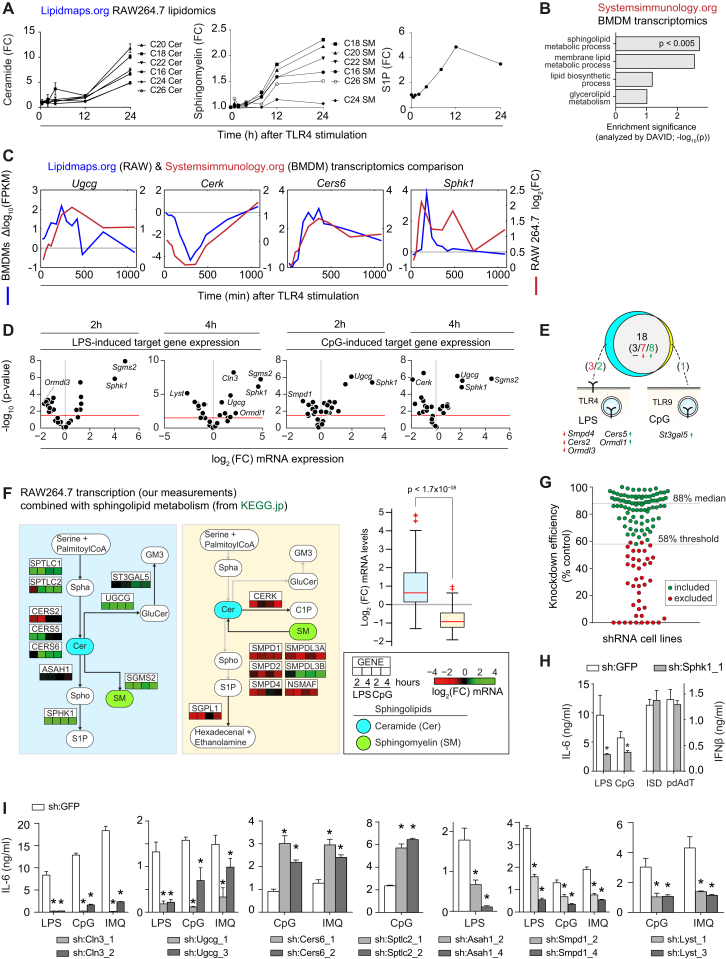
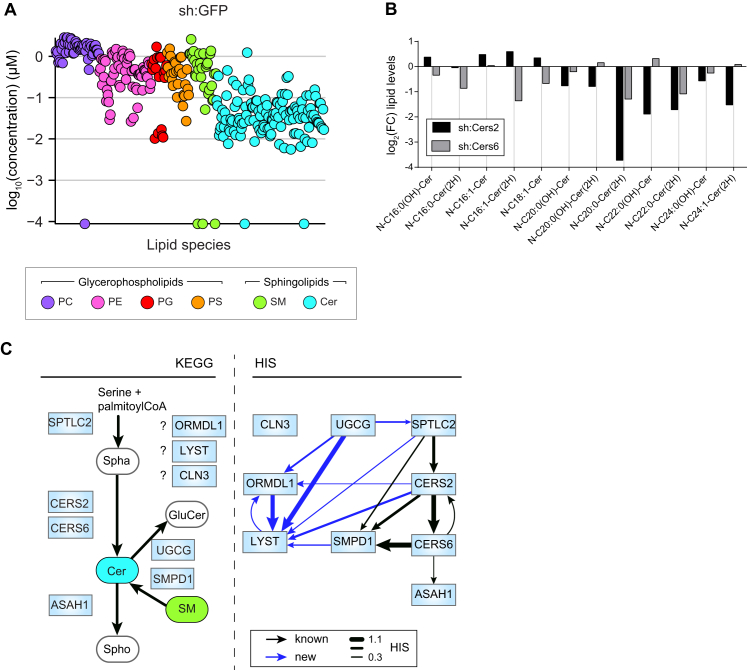
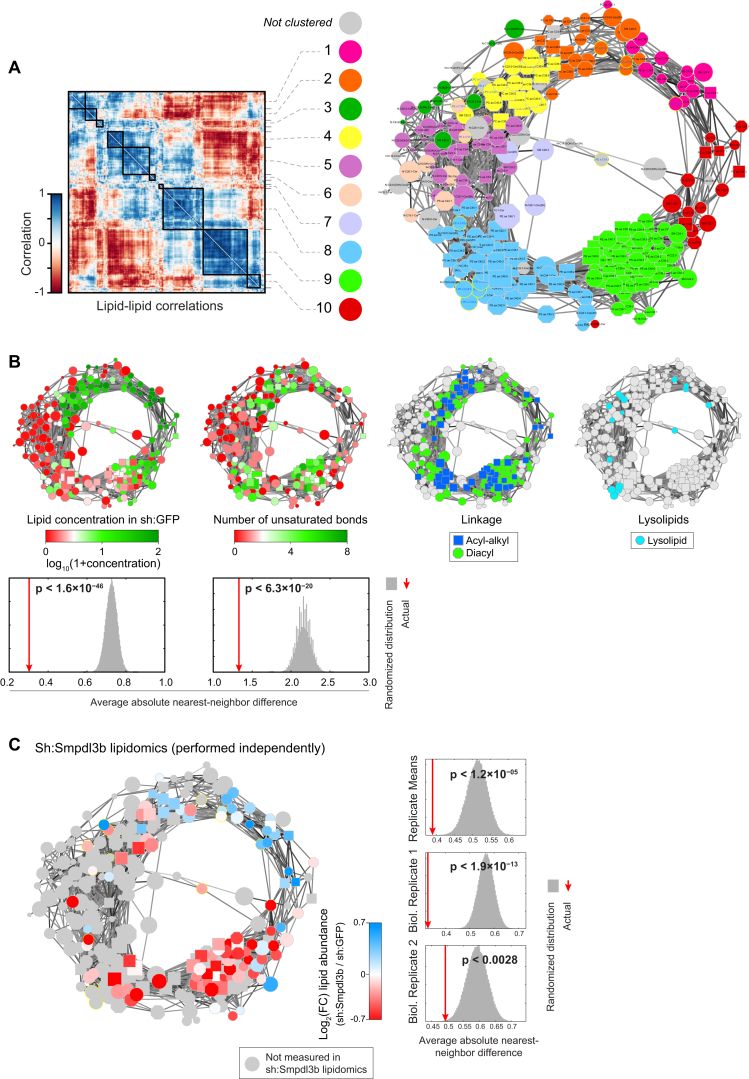
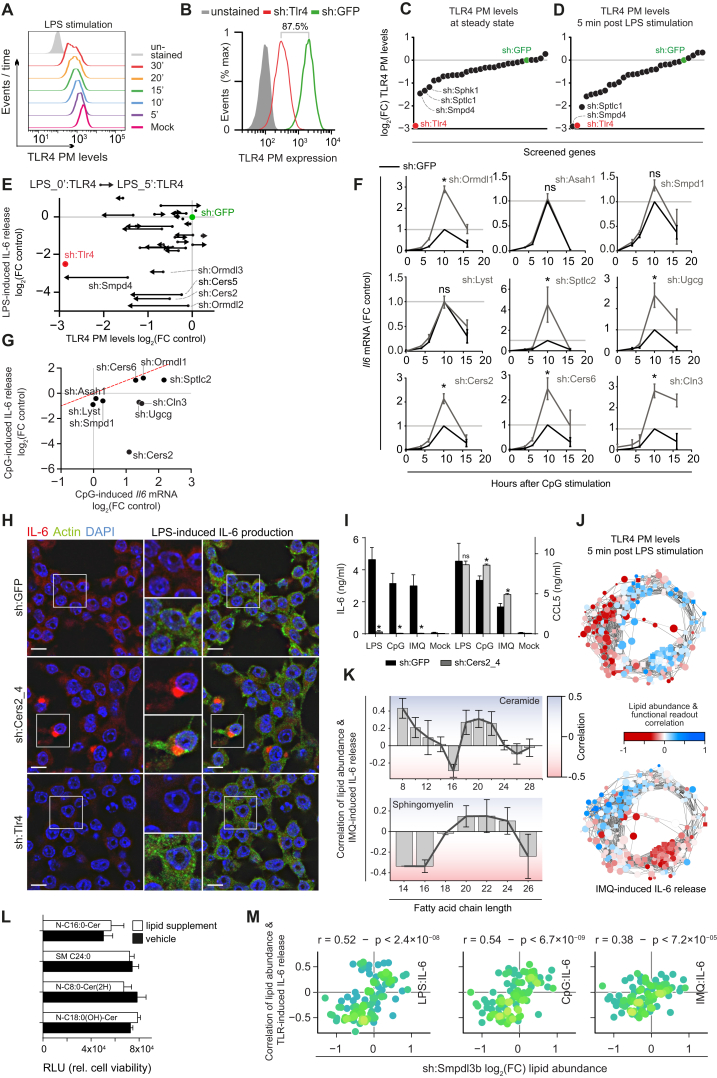
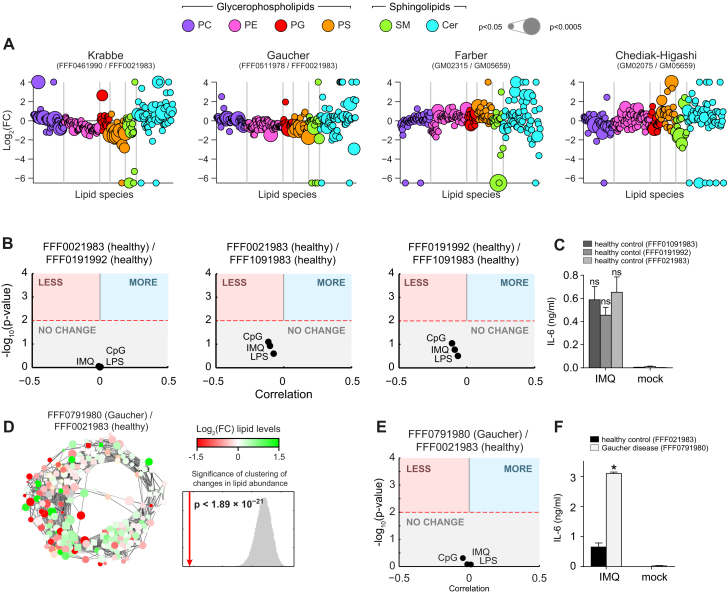
Lipid composition affects the biophysical properties of membranes that provide a platform for receptor-mediated cellular signaling. To study the regulatory role of membrane lipid composition, we combined genetic perturbations of sphingolipid metabolism with the quantification of diverse steps in Toll-like receptor (TLR) signaling and mass spectrometry-based lipidomics. Membrane lipid composition was broadly affected by these perturbations, revealing a circular network of coregulated sphingolipids and glycerophospholipids. This evolutionarily conserved network architecture simultaneously reflected membrane lipid metabolism, subcellular localization, and adaptation mechanisms. Integration of the diverse TLR-induced inflammatory phenotypes with changes in lipid abundance assigned distinct functional roles to individual lipid species organized across the network. This functional annotation accurately predicted the inflammatory response of cells derived from patients suffering from lipid storage disorders, based solely on their altered membrane lipid composition. The analytical strategy described here empowers the understanding of higher-level organization of membrane lipid function in diverse biological systems.
Copyright © 2015 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Barak V., Acker M., Nisman B., Kalickman I., Abrahamov A., Zimran A., Yatziv S. Cytokines in Gaucher’s disease. Eur. Cytokine Netw. 1999;10:205–210. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
