

Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis
- PMID: 26095251
- PMCID: PMC4508757
- DOI: 10.1016/j.cell.2015.05.047
Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis
Abstract
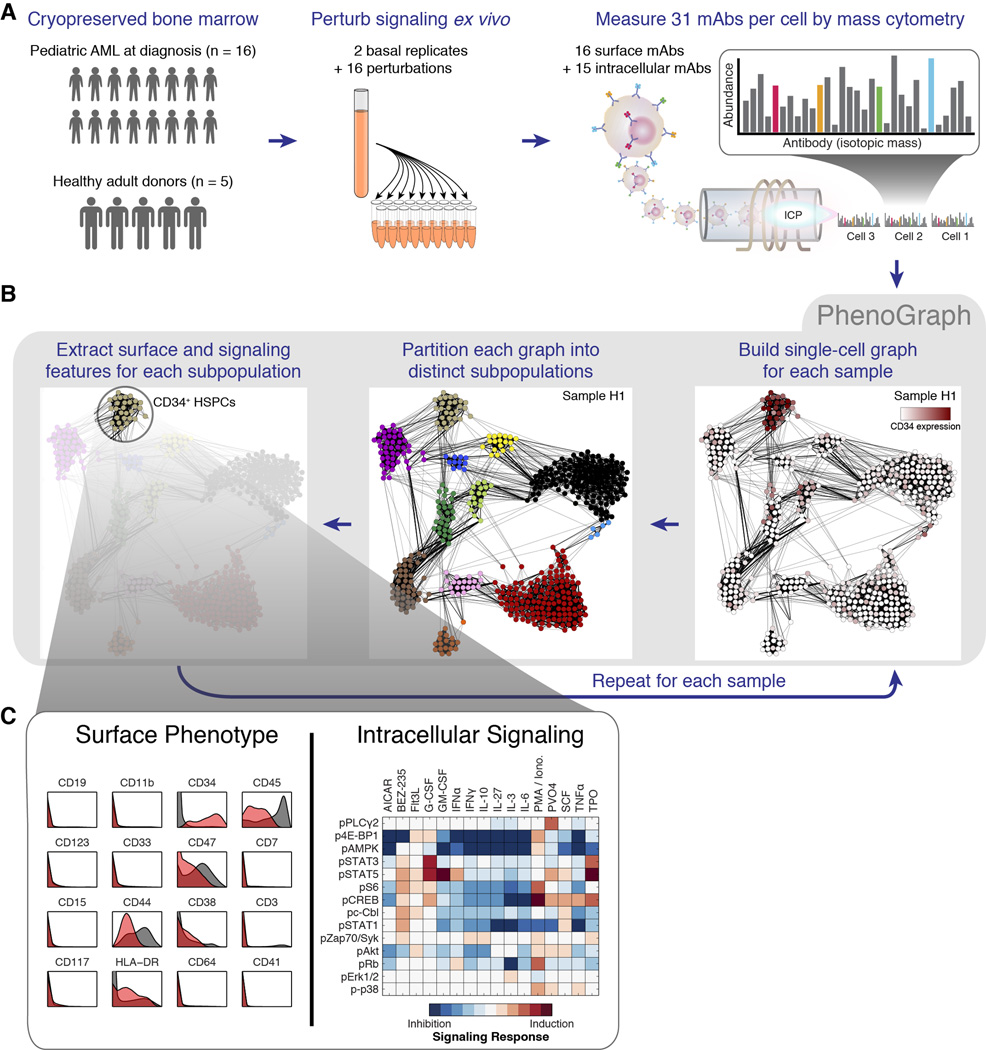
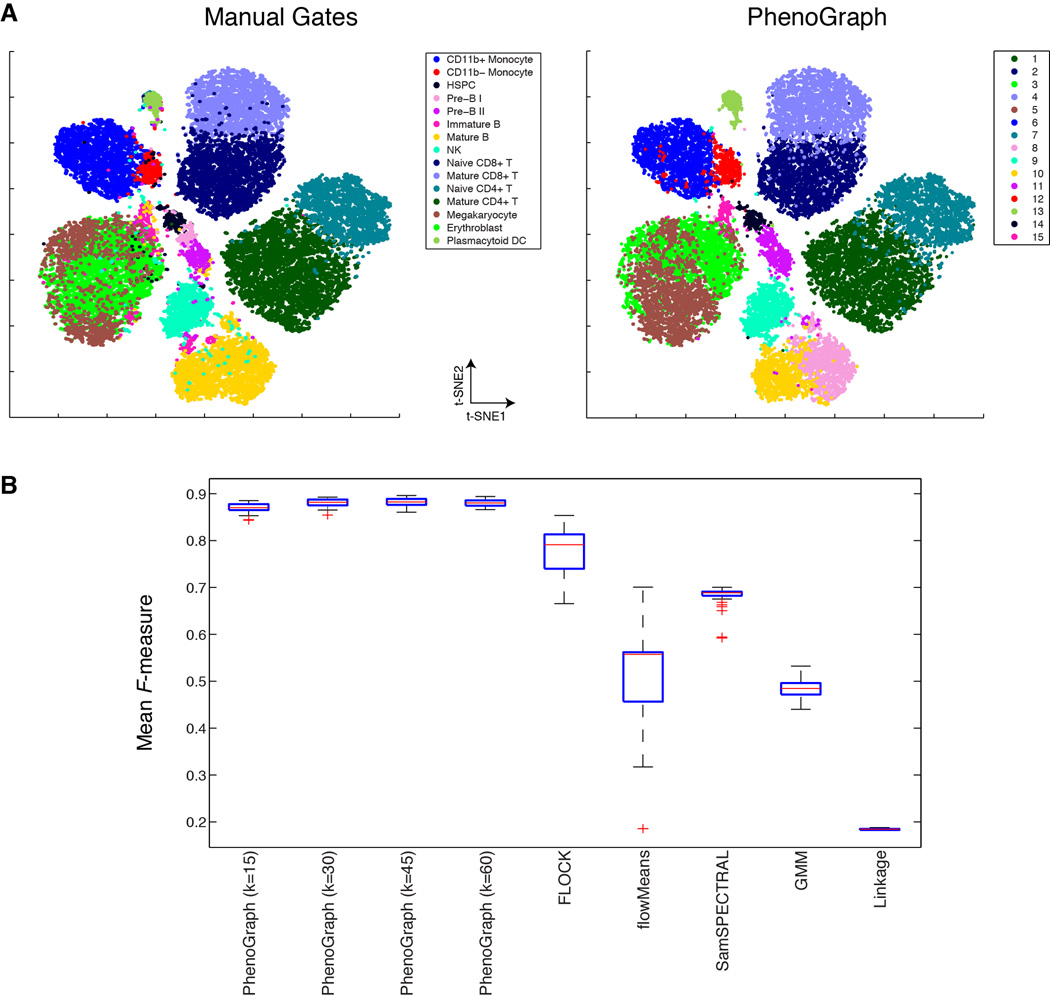
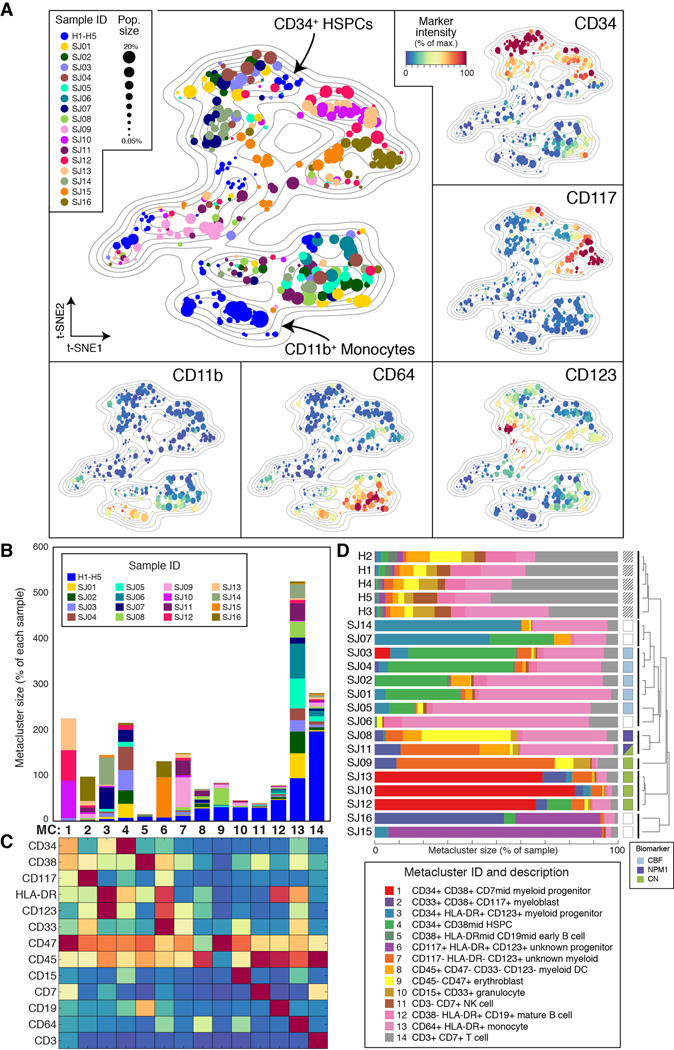
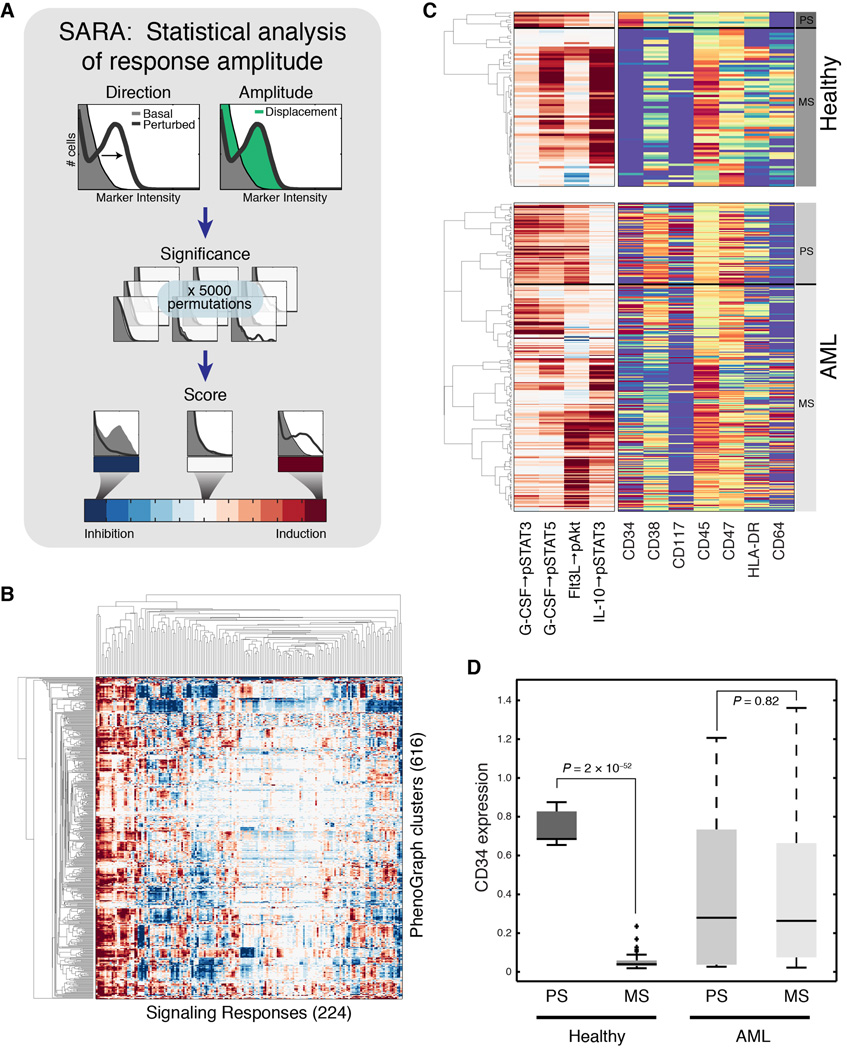
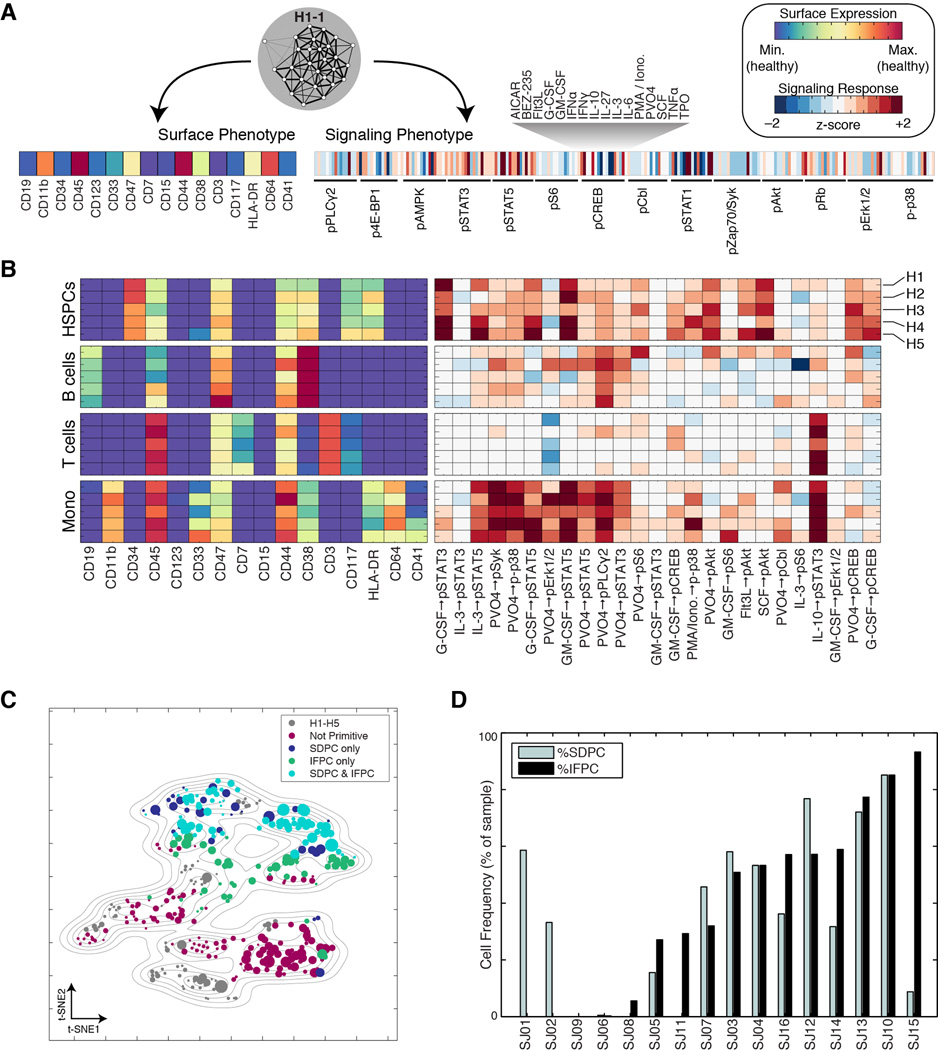
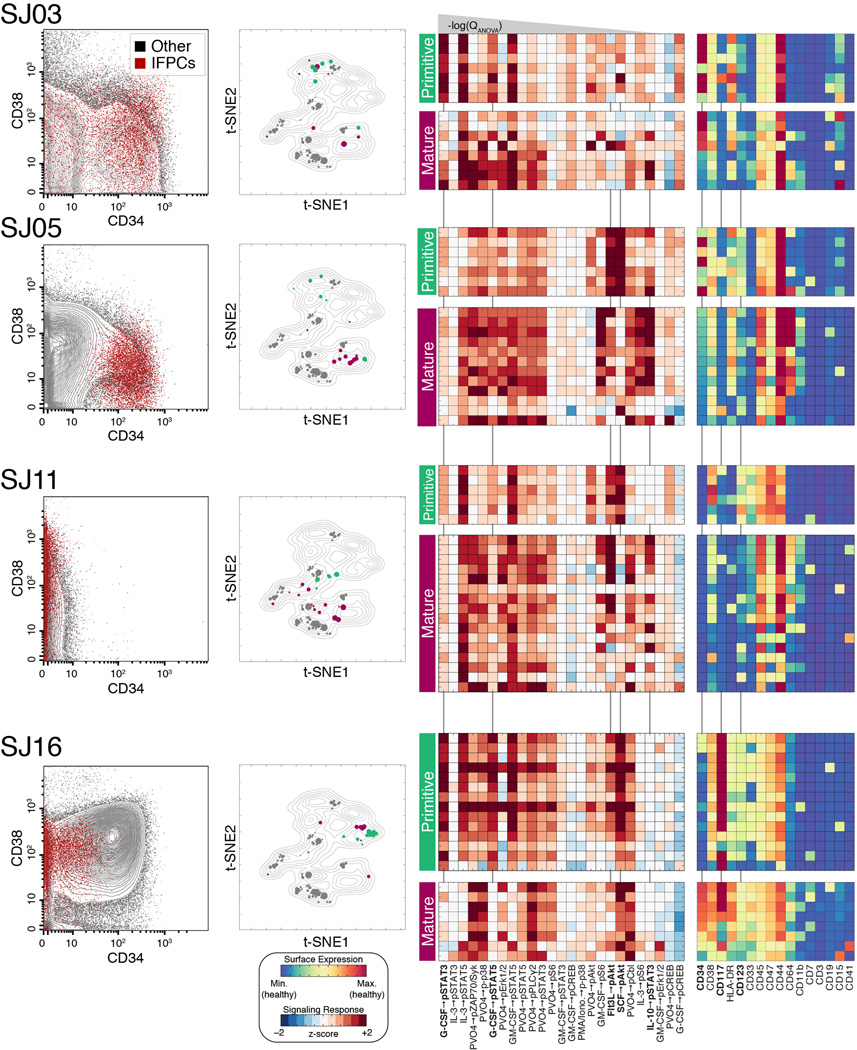
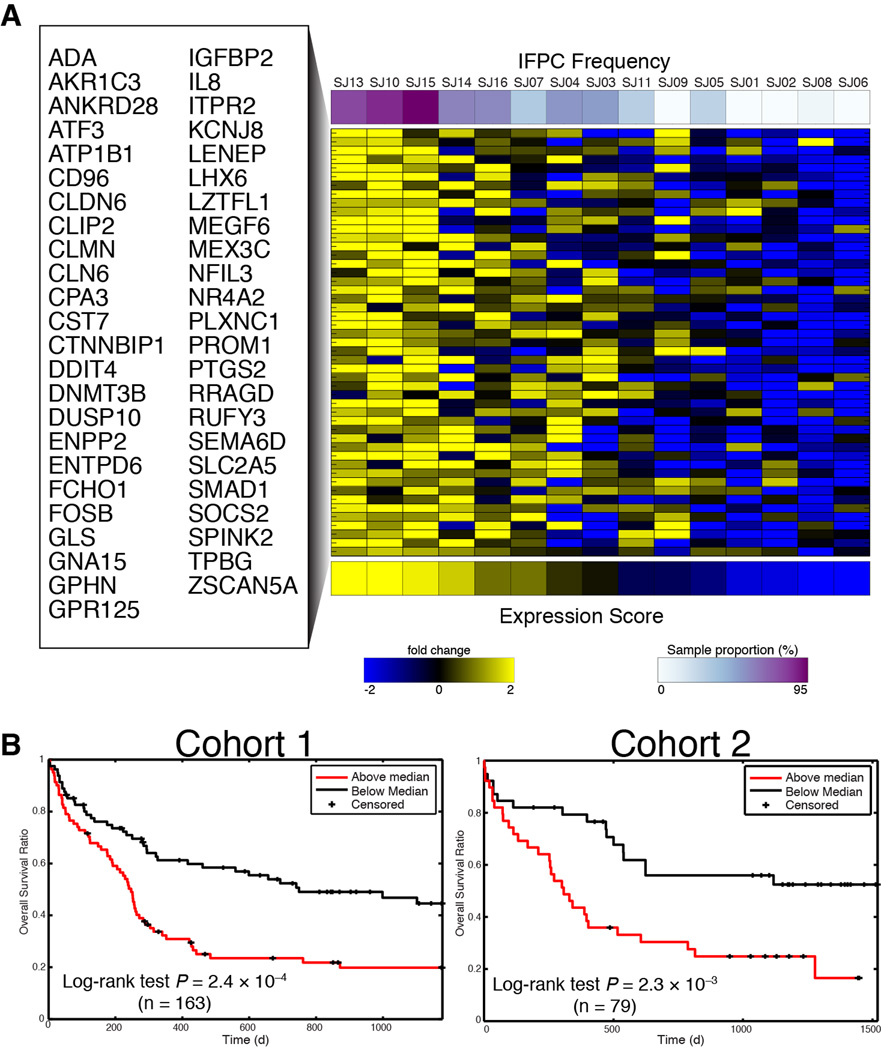
Acute myeloid leukemia (AML) manifests as phenotypically and functionally diverse cells, often within the same patient. Intratumor phenotypic and functional heterogeneity have been linked primarily by physical sorting experiments, which assume that functionally distinct subpopulations can be prospectively isolated by surface phenotypes. This assumption has proven problematic, and we therefore developed a data-driven approach. Using mass cytometry, we profiled surface and intracellular signaling proteins simultaneously in millions of healthy and leukemic cells. We developed PhenoGraph, which algorithmically defines phenotypes in high-dimensional single-cell data. PhenoGraph revealed that the surface phenotypes of leukemic blasts do not necessarily reflect their intracellular state. Using hematopoietic progenitors, we defined a signaling-based measure of cellular phenotype, which led to isolation of a gene expression signature that was predictive of survival in independent cohorts. This study presents new methods for large-scale analysis of single-cell heterogeneity and demonstrates their utility, yielding insights into AML pathophysiology.
Copyright © 2015 Elsevier Inc. All rights reserved.
Figures
Comment in
-
From mass cytometry to cancer prognosis.Nat Biotechnol. 2015 Sep;33(9):931-2. doi: 10.1038/nbt.3346. Nat Biotechnol. 2015. PMID: 26348963 No abstract available.
References
Publication types
MeSH terms
Grants and funding
- R01 CA164729/CA/NCI NIH HHS/United States
- U54-CA121852/CA/NCI NIH HHS/United States
- R01-CA164729/CA/NCI NIH HHS/United States
- U54 CA143907/CA/NCI NIH HHS/United States
- DP2-OD002414/OD/NIH HHS/United States
- T32 GM008798/GM/NIGMS NIH HHS/United States
- 5U54CA143907/CA/NCI NIH HHS/United States
- U54 CA149145/CA/NCI NIH HHS/United States
- HHSN272200700038C/AI/NIAID NIH HHS/United States
- R00 GM104148/GM/NIGMS NIH HHS/United States
- DP1-HD084071/DP/NCCDPHP CDC HHS/United States
- HHSF223201210194C/PHS HHS/United States
- R01 CA130826/CA/NCI NIH HHS/United States
- HHSN272200700038C/AI/NIAID NIH HHS/United States
- P01 CA034233/CA/NCI NIH HHS/United States
- 1R00-GM104148/GM/NIGMS NIH HHS/United States
- U19 AI057229/AI/NIAID NIH HHS/United States
- DP2 OD002414/OD/NIH HHS/United States
- N01-HV-00242/HV/NHLBI NIH HHS/United States
- T32 AI007328/AI/NIAID NIH HHS/United States
- DP1 HD084071/HD/NICHD NIH HHS/United States
- U54CA149145/CA/NCI NIH HHS/United States
- 1R01CA130826/CA/NCI NIH HHS/United States
- U54 CA121852/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
