

Subclonal diversification of primary breast cancer revealed by multiregion sequencing
- PMID: 26099045
- PMCID: PMC4500826
- DOI: 10.1038/nm.3886
Subclonal diversification of primary breast cancer revealed by multiregion sequencing
Abstract
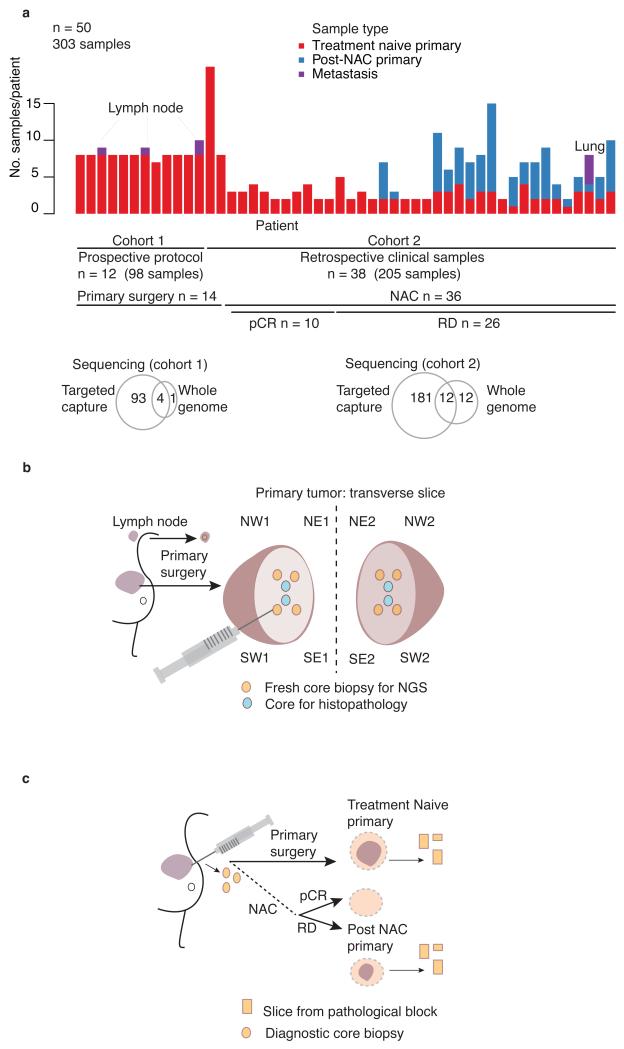
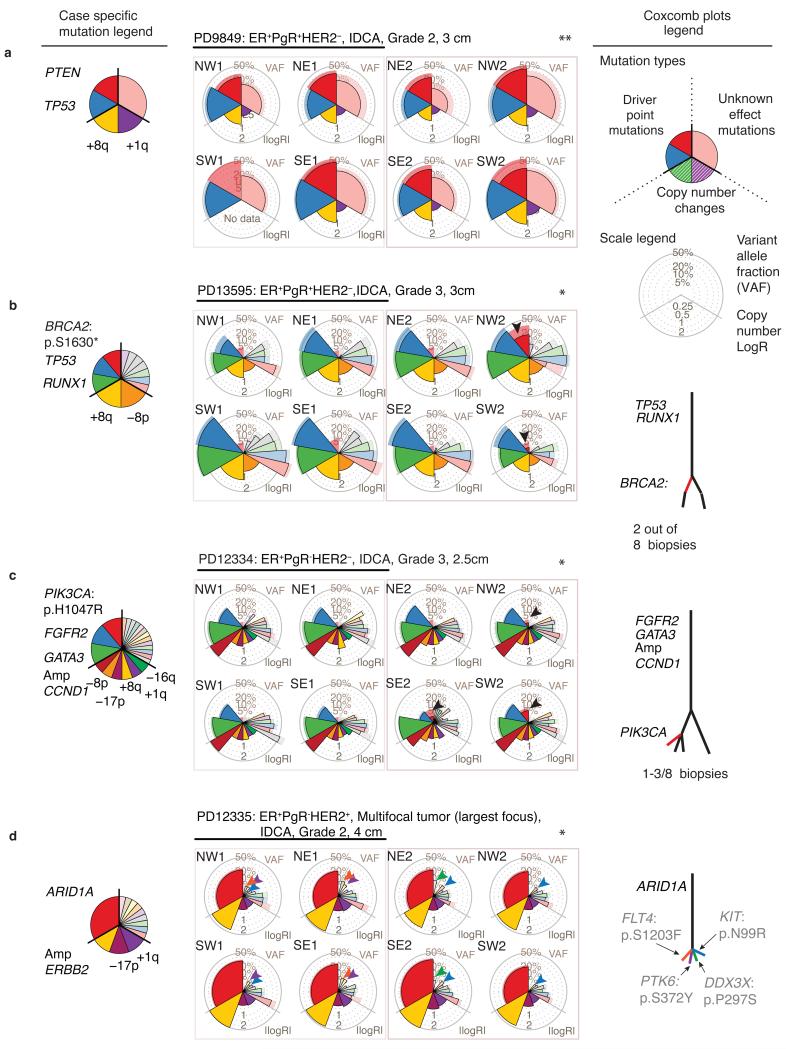
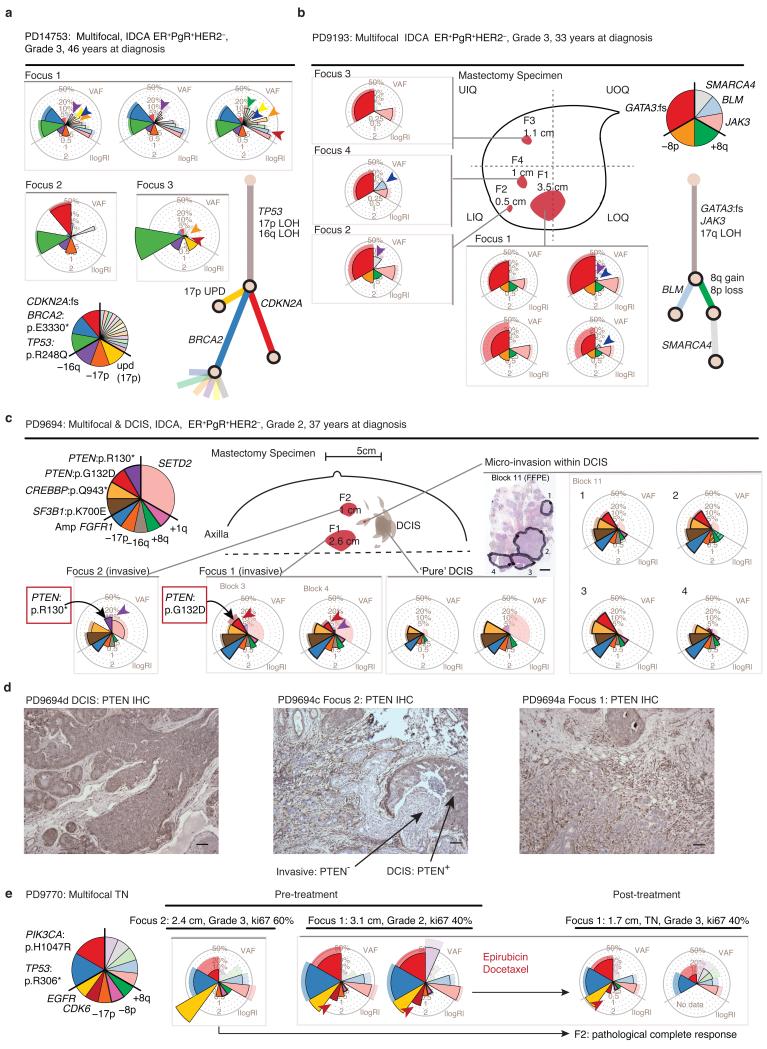
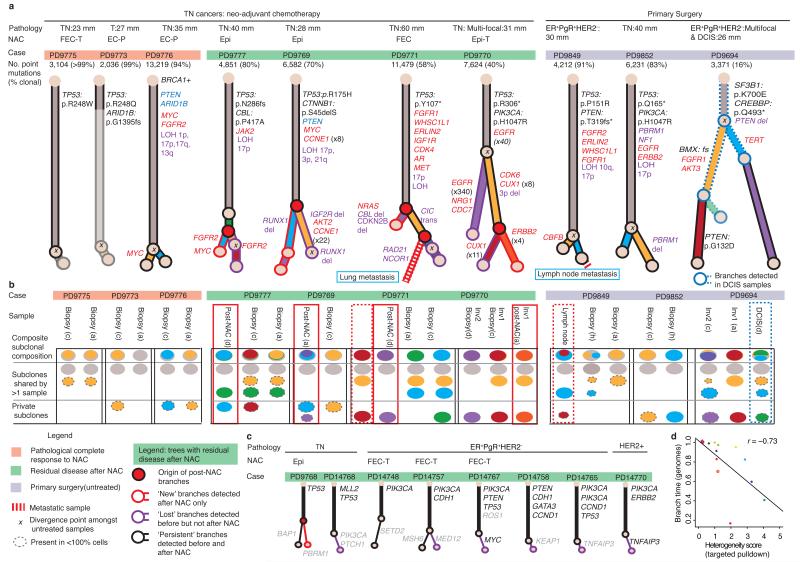
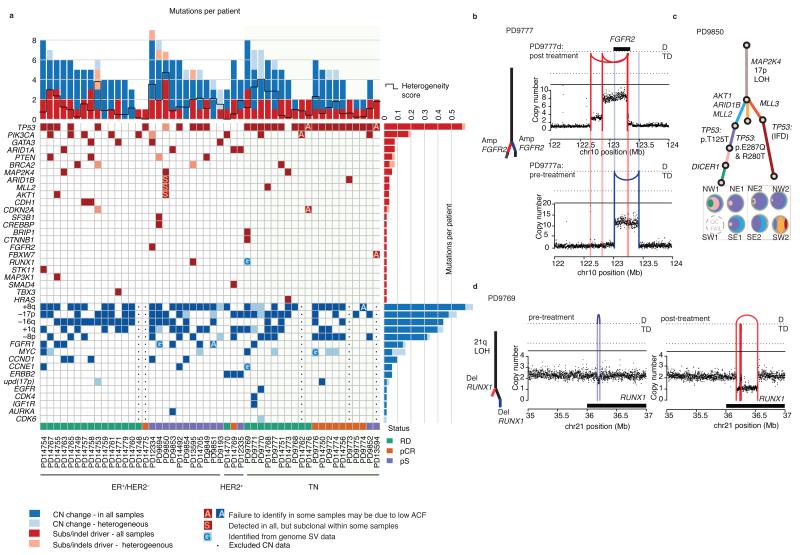
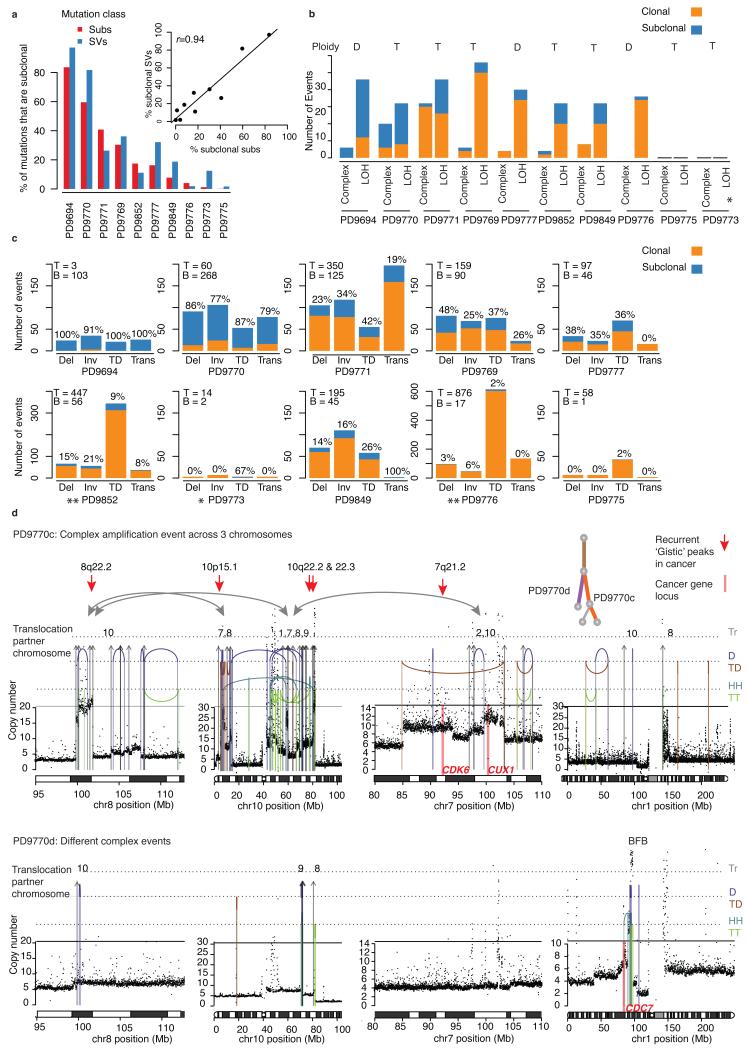
The sequencing of cancer genomes may enable tailoring of therapeutics to the underlying biological abnormalities driving a particular patient's tumor. However, sequencing-based strategies rely heavily on representative sampling of tumors. To understand the subclonal structure of primary breast cancer, we applied whole-genome and targeted sequencing to multiple samples from each of 50 patients' tumors (303 samples in total). The extent of subclonal diversification varied among cases and followed spatial patterns. No strict temporal order was evident, with point mutations and rearrangements affecting the most common breast cancer genes, including PIK3CA, TP53, PTEN, BRCA2 and MYC, occurring early in some tumors and late in others. In 13 out of 50 cancers, potentially targetable mutations were subclonal. Landmarks of disease progression, such as resistance to chemotherapy and the acquisition of invasive or metastatic potential, arose within detectable subclones of antecedent lesions. These findings highlight the importance of including analyses of subclonal structure and tumor evolution in clinical trials of primary breast cancer.
Figures
References
Online methods references
-
- Denkert C, et al. Tumor-associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010;28:105–113. - PubMed
-
- Supek F, Minana B, Valcarcel J, Gabaldon T, Lehner B. Synonymous mutations frequently act as driver mutations in human cancers. Cell. 2014;156:1324–1335. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
